与其他高通量测序技术类似,10x Genomics单细胞转录组测序数据的各项主要指标需要经过统计和质控,以便评价测序数据的整体质量。在众多的指标中,关于测序饱和度(sequencing saturation)流传着多种说法:“饱和度高于70%数据才够用”,“增加数据量就能增加饱和度”,“增加捕获细胞数必须增加测序数据量才能保证测序饱和度”……这些言之凿凿的说法,有时候却彼此矛盾,给10x Genomics技术的初学者带来了不少困扰。
饱和度作为质控标准是否如此重要?饱和度不高又需要如何优化?今天,小编整理了10x Genomics官方说明和大量安诺项目经验,为你揭开10x Genomics单细胞转录组“测序饱和度”的神秘面纱~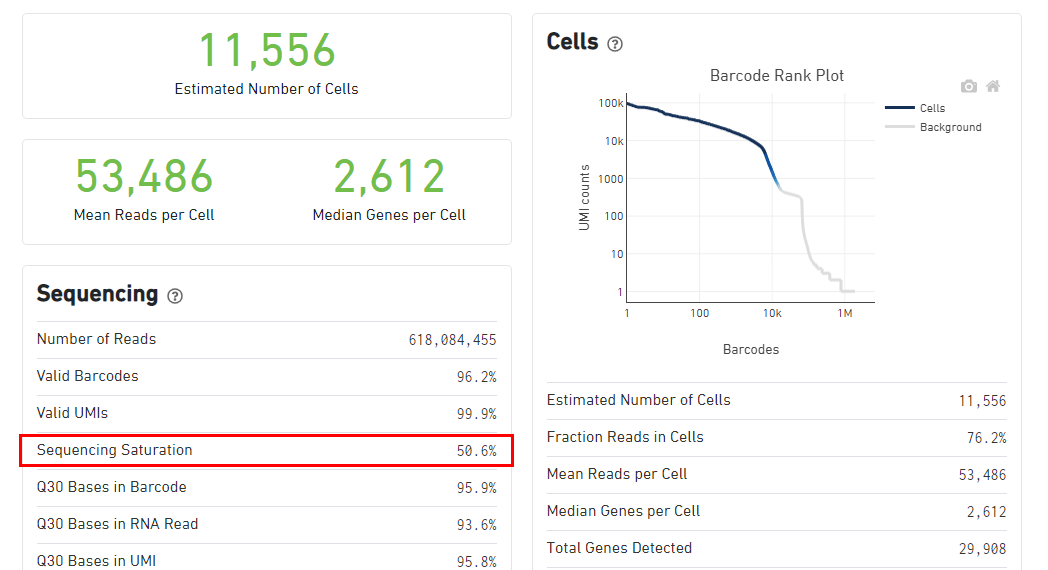
Cell Ranger分析结果展示,红色方框中就是今天的主角!
1. 什么是测序饱和度?
测序饱和度是反映当前测序量与文库复杂度相关性的指标,其大小主要取决于测序深度和文库复杂度。
测序饱和度受测序深度影响。一般来说,测序reads越多,被检测到的独特转录本就越多。如下图所示,被检测到的基因数会随着测序深度的增加而增加,但当测序深度达到一定程度,被检测到基因数量的增加程度逐渐变缓,直到不再随测序深度增加而增加即达到饱和。最终可检测到的基因数量取决于细胞类型。
测序饱和度还受文库复杂度的限制。通常来说,不同类型的细胞含有不同数量、不同类型的RNA,因此不同类型细胞建成的文库中也包含着不同数量、不同类型的转录本,即文库复杂度存在差异。复杂度高的文库中转录本的数量和类型更多,检测一个新转录本所需的额外reads数量也就更多,即增加饱和度需要测定的reads更多。
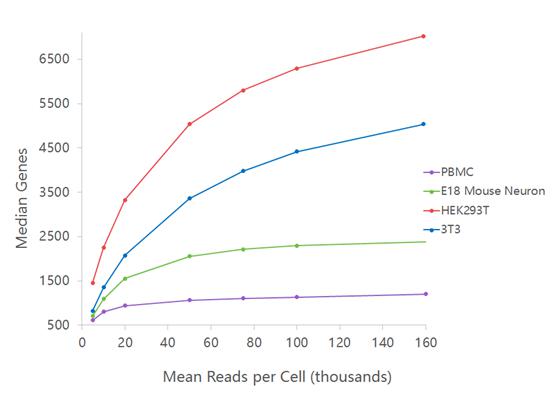
10x Genomics单细胞转录组文库测序深度与每细胞检测到基因中位数的关系图
注:原代细胞类型(如PBMC和小鼠胚胎神经元) 转录本类型少,相同reads数量下检测到的转录本类型少,且较少reads数量就能达到饱和;细胞系(如 hek293t 和3t3)转录本类型多,相同reads数量下测到的转录本类型多,很难达到饱和。
2. 测序饱和度是如何算出来的呢?
测序饱和度是通过计算含有有效barcode和有效UMI的非唯一reads在精确比对reads中占比得到的,其计算公式如下:Sequencing Saturation = 1 - (n_deduped_reads / N_reads)n_deduped_reads:准确比对的reads中唯一(有效barcode+有效UMI+基因)组合的数量N_reads:准确比对的reads中所有(有效barcode+有效UMI+基因)组合的数量简单来说,在数据准确有效的情况下,每检测到一种独特的reads,该项目的reads类型计数增加1,N_reads表示该项目共检测到了N种独特的reads。n_deduped_reads表示在N种独特的reads之中,有n种reads仅被检测到了1次。测序饱和度是指至少被检测到2次的reads占比,也就是1 - (n_deduped_reads / N_reads)。3. 测序饱和度多少才合适呢?
测序饱和度是衡量测序文库复杂度的指标。是否需要高测序饱和度,取决于实验的目的。如果研究目的是将细胞聚集成不同亚群后进行下游分析,检测的对象主要是在细胞中高表达的标志基因(marker genes),那么就不需要检测到每个细胞中所有独特的转录本,较低的测序饱和度就可以满足该研究目的。如果试图鉴定各细胞中低表达的转录本,则需要更高的测序饱和度。测序饱和度的倒数可以解释为检测一个新转录所需的额外reads。如果测序饱和度是50% ,那么概率上每多测2个reads将有一个新的 UMI(唯一的转录本)计数;相比之下,测序饱和度达到90% 时,意味着需要多测10个reads才能获得一个新的UMI(唯一的转录本)计数。如果测序饱和度很高,额外的测序将不会获得更多有效信息。
