

最近在处理一批RNA-seq的数据,里面混入了spike-in。利用spike-in矫正之后,样本A的基因表达量普遍比样本B的基因表达量高3-5倍,这和我所熟知的背景知识是一致的。
但是当我使用DESeq2对基因表达量进行差异表达分析时,上调的基因和下调的基因竟然相差无几,都有两三千个……这不符合逻辑啊,前前后后思考了一遍,发现是我对DESeq2的理解太浅的缘故。
太长不看系列
spike-in是已知的其他物种基因组,可以对基因表达数据进行绝对值的矫正
个人认为,FPKM/RPKM/TPM都是基因的相对表达值
DESeq2会对测序深度进行矫正,将普遍高表达的样本A看作是测序深度过高所导致,从而影响差异基因的筛选
废话超多系列
RNA的spike-in是一组序列已知的RNA转录本,目前普遍使用的是ERCC( The External RNA Control Consortium)搞出来的一组RNA序列。当然,也可以使用序列相似度较低的物种的序列作为spike-in。如两种常见的酵母,S.pombe和S. cerevisiae,他们的序列可以作为彼此的spike-in进行后续矫正。
通过在样品制备过程中,混入指定数量的spike-in,我们就可以知道不同样本中的基因绝对比表达值。
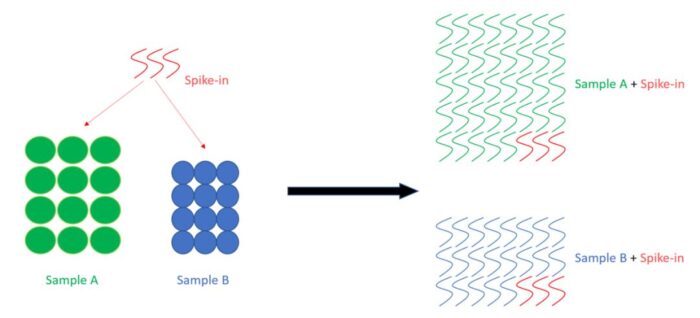
如等细胞数的样本A和样本B,在每个样本中,我加入了等量的spike-in。最后分析发现,spike-in占样本A的1%,但是占样本B的5%。这表明样本A的RNA表达量也许普遍比样本B的表达量高五倍左右。
下面是一个简单的草图,希望可以帮助理解,左边是细胞,右边是RNA
说完了spike-in矫正的原理,接下来讲讲DESeq2文库矫正的原理。
常用的normalization方法有FPKM/RPKM/TPM,但是这些方法只能对测序深度和基因长度进行矫正,没有考虑样本的文库组成成分可能对表达量产生的影响。这里举一个例子:

看起来两个不同的样本之间除了基因A1CF,其余基因都是差异表达的。但事实上,这是由于样本A中A2M表达量过高,导致样本中其他基因的相对表达量较低。如果我们把两个样本中的A2M基因去除,重新计算FPKM,我们会发现两个样本中其他五个基因的表达量其实相差无几。
因此DESeq2需要解决两个问题:
1、样本的测序深度
2、样本的文库组成
这里以一个简单的例子讲解一下DESeq2是如何解决这两个问题的。
首先对样本量进行以自然对数为底数的log转换:

DESeq2除了可以使用,还可以使用和,但因为R默认的log是以e为底数,因此这里使用。我曾经画图时为了偷懒使用,导致组会上被老板批评,因为搞生信的前辈们普遍喜欢使用或者进行对数转换。我太难了╥﹏╥...
注:我们可以看到,表达量为0的值在去对数之后,变成了负无穷。
对每一行的值进行平均值计算

第二列和第三列都比较好理解,第一列因为样本1的gene1的log值是-inf,因此gene1这一列的平均值也是-inf。这里还有一点值得关注,当我们将gene3的平均表达的log转换为正常数值时,≈73.7,而对基因表达值的原始矩阵计算得到的平均值为(33 55 200)/3=96。
我们可以看到96>73.7,因此这种取对数的方法可以使得基因更不容易受到异常值的影响。事实上这种取完log之后做平均的方法,计算的是几何平均数
移除具有-inf值的基因
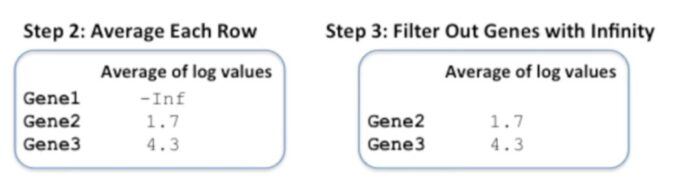
当一个样本或者多个样本中基因的表达量为0时,这个基因就会被移除。
事实上,通过这个可以让我们最后的基因表达矩阵更加集中于管家基因(house-keeping gene)
对基因的reads count取log并减去基因的log值的平均数,具体如下:

通过这个计算方式,我们将得到每个样本中geneX的表达水平与 geneX在所有样本平均表达水平的比值
计算步骤四所得的样本表达矩阵中,每个样本中的基因表达中位数
这里使用中位数,可以进一步减少异常值对于scale factor(校正因子)的影响。至于为什么中位数有这个好处,我想这里应该不需要详细阐述了

将步骤5计算的每个样本的中位数,进行指数计算

通过该方法,我们最终得到了样本的校正因子(scale factor)
将原始样本表达矩阵除以步骤6所得到的scale factor

以上七步就是DESeq2对于样本文库矫正的方法,
为避免文章太长,对前文有所遗忘。我们在这里再简单的叙述一下,DESeq2对于样本文库的矫正做了些什么:
1、使用log转换,去除那些只在某几个样本中表达的基因,也减少了极端值对于最终结果的影响
2、取每个样本的中位数,进一步减少极端值的影响,使得结果更加关注于在多个样本中稳定表达的基因
通过对上述DESeq2的文库normalization方法的学习,我了解到我的RNA-seq数据是受到了DESeq2自动文库矫正的影响。这个是可以关闭的
这可真是个bug啊,DESeq2明明是好心却办出错了事,无奈我只能使用logFoldChange和T检验的方法筛选差异表达基因……
作者:小白要变大神
链接:https://www.jianshu.com/p/c5473644e89f
来源:简书
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。