

cell ranger输出结果目录结构如下所示
- ├── analysis
- │ ├── clustering
- │ ├── diffexp
- │ ├── pca
- │ └── tsne
- ├── cloupe.cloupe
- ├── filtered_feature_bc_matrix.h5
- ├── filtered_gene_bc_matrices
- │ └── GRCh38
- ├── metrics_summary.csv
- ├── molecule_info.h5
- ├── possorted_genome_bam.bam
- ├── possorted_genome_bam.bam.bai
- ├── raw_feature_bc_matrix
- │ ├── barcodes.tsv.gz
- │ ├── features.tsv.gz
- │ └── matrix.mtx.gz
- ├── raw_feature_bc_matrix.h5
- └── web_summary.html
输出文件非常的多,为了方便查看结果,提供了一个所有结果汇总的html页面,即web_summary.html。该网页的结果分成了summary和analysis两部分, summary部分包含如下结果
1. 异常结果警告
如果数据中存在异常,在网页的头部会给黄色的警告框,如下所示
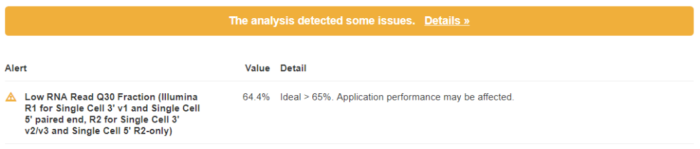
点击Details, 可以看到详细的信息,上图显示RNA reads的Q30比例太低,理想情况是大于65%, 而实际的数据只有64.4%。
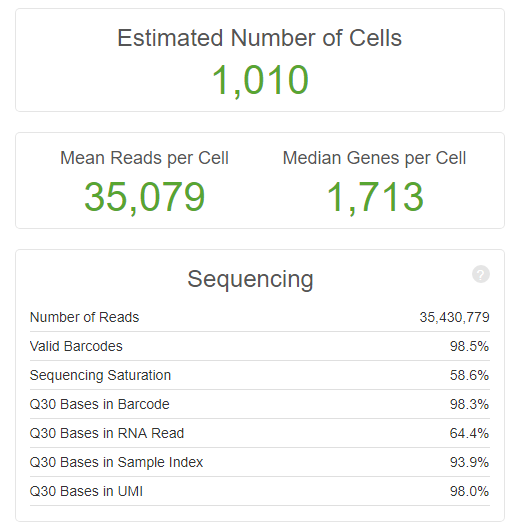
2. 细胞和基因数目的评估
对样本中的细胞和表达的基因个数评估,同时还给出了barcode, index, umi, RNA reads不同序列的Q30, 示意如下
3. 比对比例统计
统计reads的比对比例,同时给出比对到基因间区,外显子,内含子的比例,示意如下

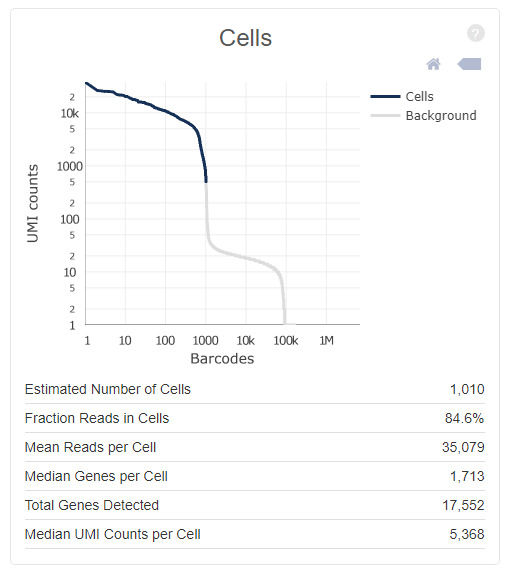
4. 细胞数目评估详细信息
通过barcode上的UMI标签分布来评估细胞数目,深蓝色代表细胞,灰色代表背景,示意如下
5. 样本基本信息
样本名称,使用的参考基因组等信息,示意如下

analysis部分包含如下结果
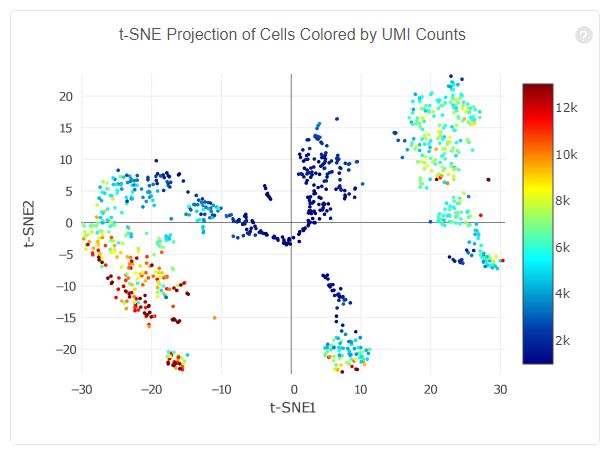
1. 细胞表达量分布的t-SNE图
UMI标签用于标识转录本,UMI的count值就是转录本的表达量,采用tSNE降维算法, 对细胞的表达量进行可视化,每个点代表一个细胞,示意如下
2. 细胞亚型
根据表达量对细胞进行聚类,从而识别细胞亚型,提供了两种聚类算法graph-based和k-means, 示意如下
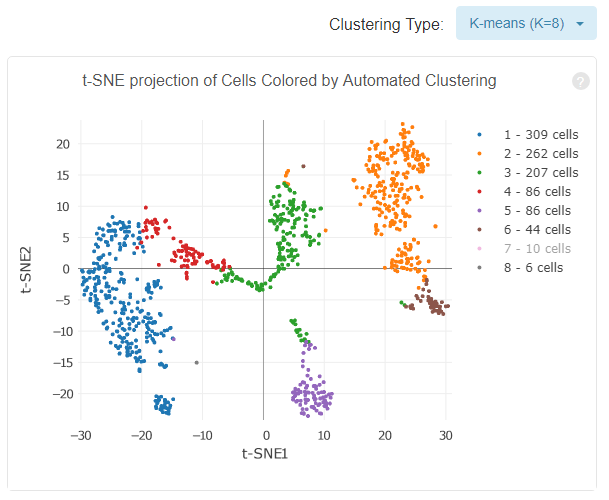
通过右上角的下拉按钮,可以查看不同的聚类结果,结果展示依然是用的t-SNE图,只不过根据聚类结果对颜色进行了调整,属于同一类的细胞用相同颜色表示。
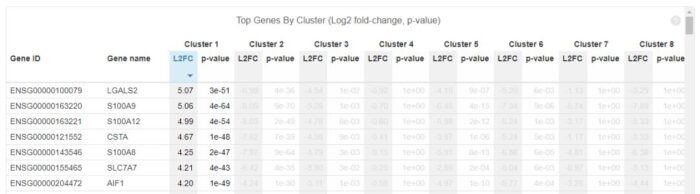
3. 基因差异表达分析
对cluster下的基因进行差异分析,将细胞分成了该cluster和其他cluster两类,然后进行差异分析,结果如下所示
4. 饱和度评估
对reads抽样,观察不同抽样条件下检测到的转录本数量占检测到的所有转录本的比例,并绘制如下曲线
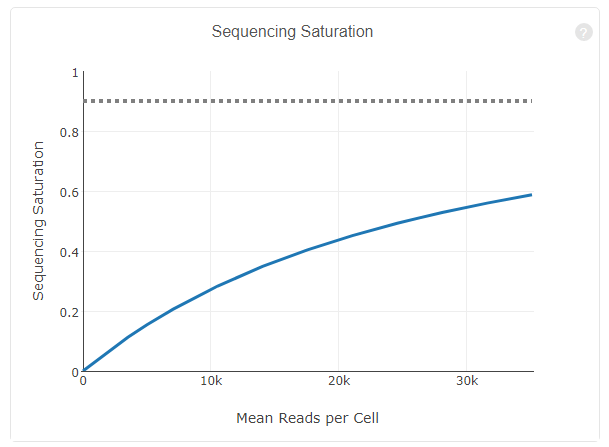
如果曲线末端区域平滑,说明测序接近饱和,再增加测序量,覆盖到的转录本数目也不会变化太多。
对reads抽样,观察不同测序数据量情况下检测到的基因数目的分布,并绘制如下曲线
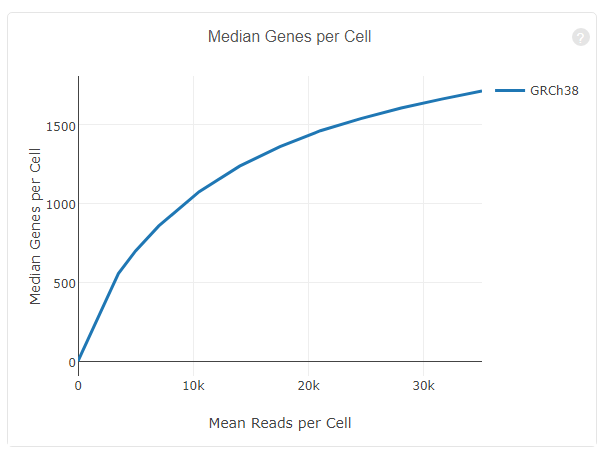
如果曲线末端区域平滑,说明测序接近饱和,再增加测序量,检测到的基因数目也不会变化太多。
网页提供了一个简单直观的查看结果的方式,但是很多细节还是需要到对应的文件中进行查找。网页中summary部分的结果保存在metrics_summary.csv文件中,analysis部分结果保存在名为analysis的文件夹下,该文件夹包含以下4个子目录
- ├── clustering
- ├── diffexp
- ├── pca
- └── tsne
pca是表达量的PCA分析结果,tsne是表达来量的t-SNE分析结果,diffexp是差异分析的结果,clustering是聚类的结果,每个聚类结果都提供了一个cluster.csv文件,内容示意如下
可以看到barcode和cluster的对应关系,barcode用于标识细胞,这张表格反映的是细胞和cluster的对应关系。
在结果目录,可以看到如下两个目录
- raw_feature_bc_matrix
- filtered_gene_bc_matrices
这两个目录下的内容是类似的,raw目录下是所有的barcode信息,包含了细胞相关的barcoed和背景barcode,而filter目录下只包含细胞相关的barcode信息,内容如下
- ├── barcodes.tsv
- ├── genes.tsv
- └── matrix.mtx
后缀为mtx的文件记录的就是基因的表达量信息,可以导入R或者python中查看,也可以通过如下命令转换为csv格式
- cellranger mat2csv \
- outs/filtered_gene_bc_matrices \
- sample.count.csv
除了用MEX格式来存储表达量数据,还使用用HDF5的格式来记录表达量信息,对应以下两个文件
- raw_feature_bc_matrix.h5
- filtered_feature_bc_matrix.h5
在输出目录下,还包含以下几个文件
- possorted_genome_bam.bam
- possorted_genome_bam.bam.bai
- molecule_info.h5
- cloupe.cloupe
前两个是比对产生的bam文件和索引,第三个文件是实验相关的文库,GEM,barcode表达量等信息的HDF5格式的文件,cloupe文件则是Loupe Cell Browser的输入文件,该软件是官方提供的专门用于查看数据分析结果的软件, 后续会介绍该软件的使用。