

单细胞转录组学数据能够用于推断轨迹,从而对细胞动态变化过程进行无偏倚研究。本文中,作者对45种轨迹预测方法进行了评估,并为研究者选择合适的软件方法提出了实质性的指导意见。
Introduction
单细胞数据可以用于研究细胞动态过程,如细胞周期,分化以及细胞活化等。轨迹预测(trajectory inference, TI)方法,又称拟时间分析(pseudotime analysis)方法,能够基于表达模式的相似性,将细胞沿着一条线性,分叉,或者更为复杂的树或图状结构进行排列,从而用于鉴定新的细胞亚群,描绘分化轨迹,以及推断导致分叉的调控程序等。从2014年起,有超过70种 TI 方法发布,但还未有人对这些方法进行一个系统的比较。不同的方法具有不同的特征,包括对输入的要求,输出结果的结构,以及使用的具体算法等,使得使用者难以确定到底使用哪种方法用于自己的研究,所以我们需要从各种方法的表现,扩展性,鲁棒性以及用户使用角度入手,对其进行全面的评估
方法筛选以及评价标准
本研究中共囊括了45种 TI 方法。45种方法的主要特征以及总体的评估结果如下图所示
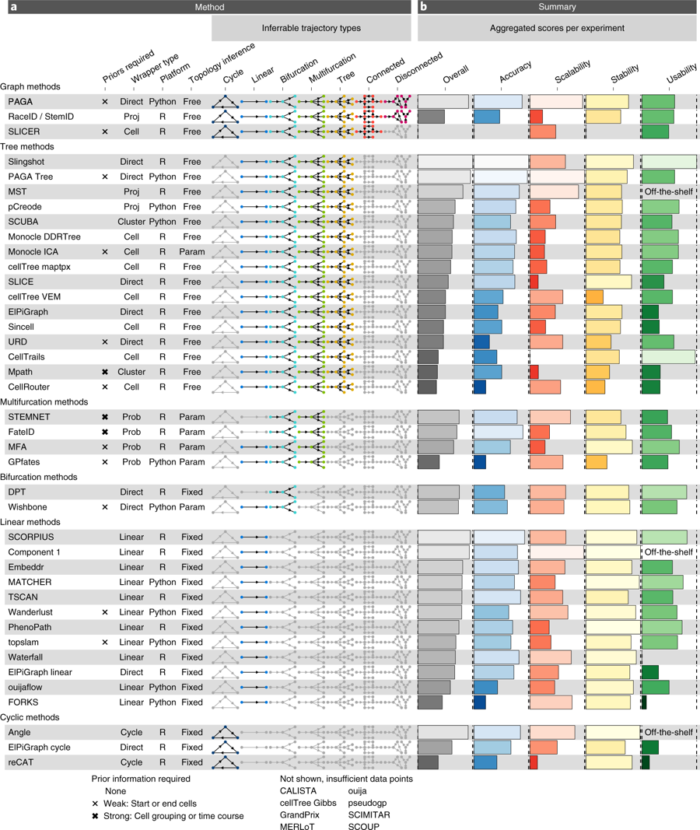
部分方法对输入数据有要求,有指定一个起始细胞的,还有需要已知细胞分组的。如果某种方法必须,则提供其所需的先验信息
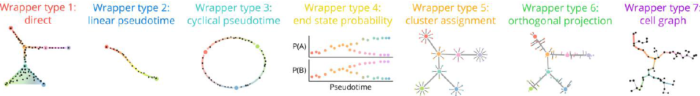
不同方法的输出结果也不同,但是能够将它们分为7大组,作者为每一组编写了通用的转换器(下图),实现对结果的封装,用于比较
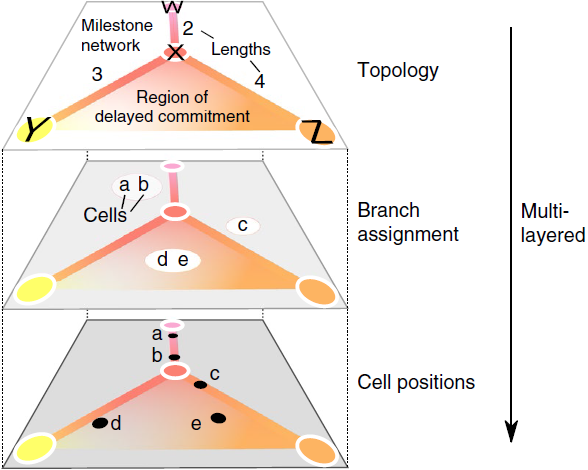
为了能够直观地比较不同方法的输出结果,作者开发了一套通用概率模型用于表示轨迹(下图)。在该模型中,整个轨迹的拓扑结构通过一个由“里程碑”构成的网络来表示,细胞则置于相连的点构成的空间中
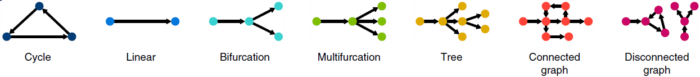
不同方法间最大的差异在于是否固定了结果的拓扑结构,如果没有的话,那么它能够检测出哪些拓扑结构。作者共定义了7种不同结构(下图)。绝大部分方法只能预测线性或其他较简单的结构,只有极少数方法能够预测环形或者分离式的拓扑结构
作者从以下4大核心层面入手评估每种方法:
- 预测的准确性(accuracy),总计110个真实数据集和229个合成数据集
- 方法的扩展性(scalability),细胞和特征(如基因)数量增加后的表现
- 预测的稳定性(stability),数据集抽样后预测的结果情况
- 工具的可用性(usability),包括软件的实现,说明文档等
具体的细则看下一部分结果
评价结果
所有方法具体的评价结果如下图所示

(各方法具体评价结果)
准确性
评估准确性使用的数据集包括两种类型:
- 真实数据集(110个),涉及不同的 scRNA-seq 技术,物种,动态过程以及拓扑结构等。如果一个真实数据集参考轨迹不是通过表达数据本身得到的,而是基于细胞分选(cell sorting)或细胞混合(cell mixing)得到,则归为 gold standard,其他的均归为 silver standard
- 合成数据集(229个),使用了多种数据模拟器,其中包括一种利用基因调控热力学模型的基因调控网络模拟器。对于每个模拟,以真实数据集作为参考,并匹配其维度,差异表达基因的数量,droupout 率以及其他统计特性
选择了4项指标(如下图所示),包括:
- 拓扑结构(topology)(Hamming–Ipsen–Mikhailov, HIM)
- 细胞分配到分支的质量(F1branches)
- 细胞的位置(cordist)
- 差异表达特征沿着轨迹分布的准确性(wcorfeatures)
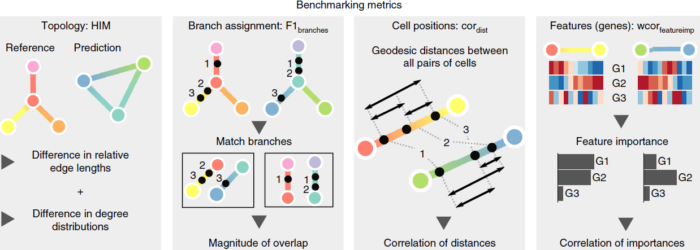
结果显示各方法在不同数据集上的表现差异较大,意味着不存在适用于所有数据集的“万全之策”。不同来源的数据集的整体分值与具有金标准的真实数据的整体分值之间存在中度到较高的相关性,表明金标准轨迹的准确性以及合成数据集之间的相关性。不同指标得到的结果常出现不一致的情况,例如 Monocle 和 PAGA Tree 在拓扑结构方面得分最高,而 Slingshot 则是在对细胞进行排序以及将其置于正确的分支方面表现更佳
方法的表现好坏高度依赖于数据集蕴含的轨迹类型。例如 Slingshot 在包含相对较简单的轨迹的数据集上表现更好,而 PAGA,pCreode 和 RaceID/StemID 则是在具有更复杂结构的数据集中表现更优(原文献 Supplementary Fig. 3c)。这一结果也反映在每种方法检测出的拓扑结构上,Slingshot 预测的结构倾向于包含更少的分支,而 PAGA,pCreode 和 Monocle DDRTree 等则是朝向更加复杂的结构(原文献 Supplementary Fig. 3d)。综上,对绝大部分方法而言,找出正确的结构是一项极具挑战性的任务
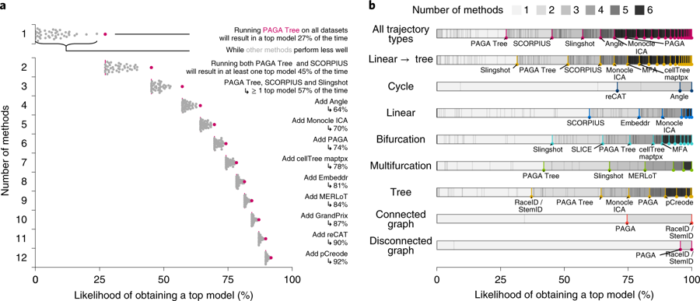
数据集之间的高度差异性以及各方法预测的拓扑结构的多样性提示可能不同方法之间存在互补性。为了验证该猜想,计算仅使用部分方法时得到 top model 的可能性,其中 top model 定义为整体得分达到最佳模型95%的模型。结果如下图a所示,仅使用单个模型时,得到 top model 的可能性为27%,而当加入另6种模型后,可能性提升至74%。分别对仅含同一种类型轨迹的数据集进行测试,结果类似,不过所需的方法数更少(下图b)。综上,用户可以考虑将多种不同的方法应用于自己的数据集,尤其是拓扑结构未知时。未来的软件开发也可以考虑利用这一点集成多类方法
扩展性
对5个不同真实数据集(测试集)放大和缩小后的版本分别测试各方法,并通过 Shape Constrained Additive Model 对运行时间和内存占用进行建模。为了验证,比较在所有339基准数据集上预测的运行时间(和占用内存)与实际值之间的相关性,结果显示整体具有极高的相关性,且具体到每种方法,也几乎都具有中到高度的相关性,意味着模型是可靠的,可用于预测各方法的扩展性
结果显示绝大部分方法的可扩展性整体而言都是较差的(上图 各方法具体评价结果 c)。面对10000 x 10000的数据集,大部分图和树方法无法在1h内完成分析;数量增加至一百万个细胞时,除 PAGA,PAGA Tree,Monocle DDRTree,Stemnet 和 GrandPrix 外,其他图/树方法无法在1d内完成分析;此外像 Monocle DDRTree 和 GrandPrix,在特征数量增加后,运行时间表现上也不尽如人意。低运行时间的方法通常具有两个典型特征:时间复杂度呈线性,以及细胞和特征的增加仅导致相对较低的时间增加,而过半的方法呈现出二次方或超二次方的复杂度,导致不适用于大型数据集
内存方面,大部分方法均能在工作站或集群(≤12GB)上运行,其中 PAGA 和 STEMNET 内存占用特别低。还有一些方法,细胞数量(RaceID/StemID, pCreode and MATCHER)或特征数量(Monocle DDRTree, SLICE and MFA)较大时,所需内存会较高
综上,数据集的维度是影响方法选择的一个重要因素,未来的方法开发也应注意这一方面的优化
稳定性
对每个基准数据集的10个不同数据集子集(95%的细胞,95%的特征)进行检测,通过比较结果相似性来评估方法的稳定性,评价标准与之前用于评价准确性的一致,结果如上图 各方法具体评价结果 d 中所示
部分方法本身或者通过参数固定了拓扑结构,理论上这样的方法结果应该更稳定,比如 SCORPIUS,MATCHER 以及 MFA 确实如此;另外一些预测结果更自由的方法,稳定性差异比较大,像 Slingshot 比 PAGA (Tree) 更稳定,而后者则比 pCreode 和 Monocle DDRTree 等更稳定
可用性
基于一系列科学和软件开发准则进行评价,包括:
- 软件打包
- 说明文档
- 自动代码测试
- 发表在同行审议期刊
此外由于安装,使用平台等原因已经筛选掉了一批软件。结果显示大部分方法都达到了基本要求,但在代码保证性和相关文档方面存在问题较多。Slingshot 和 Celltrails 的可用性得分几乎完美。可用性和方法准确性之间无明确关联
Discussion
基于上述研究,作者给出了方法选择的参考指南(下图)
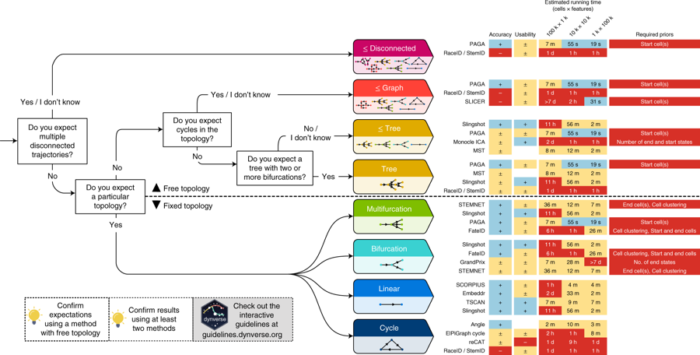
其他的建议比如在推断轨迹时,使用多种方法进行验证;即便是有预期的拓扑结构,也可以尝试其他对结构做了更少假设的方法,可能会给出新的提示
REF
- Saelens W, et al. A comparison of single-cell trajectory inference methods. Nat Biotechnol, 2019. doi: 10.1038/s41587-019-0071-9. [Epub ahead of print]
- Saelens W, et al. A comparison of single-cell trajectory inference methods: towards more accurate and robust tools. Preprint at bioRxiv https://doi.org/10.1101/276907 (2019).