

刚接触生信分析的小白们这种尴尬的事情时有发生,为了帮助大家梳理这些剪不断理还乱的文件,本文以分析流程为主线,介绍各文件的格式以及有哪些常用命令来查看或处理它们。
1. 测序数据FASTQ文件
1)文件用途:样品测序返回的数据一般存储为fastq文件,通常是压缩文件filename.fq.gz的格式,节省存储空间和传输时间。
2)查看方式
- # zcat查看gzip压缩的文件
- # head -n 8 显示前8行文件内容(前8行代表2条序列)
- zcat filename.fq.gz | head -n 8
- # @SRR1039521.13952745/1
- # TTCCTTCCTCCTCTCCCTCCCTCCCTCCTTTCTTTCTTCCTGTGGTTTTTTCCTCTCTTCTTC
- #
- # HIJIIJHGHHIJIIIJJJJJJJJJJJJJJJJJJJJJIIJJFIDHIBGHJIHHHHHHFFFFFFE
3)格式说明:fastq文件每4行代表一条序列
第一行:记录序列测序时所用仪器以及在测序通道中坐标信息,以@开头;
第二行:测序的序列信息,以ATCGN表示,由于荧光信号干扰无法判断是什么碱基时就用N表示;
第三行:通常一个 ;
第四行:与第二行碱基信息一一对应,存储测序碱基的质量值。
4)其他常用命令
- # 计算read数
- # wc -l: 计算行数
- # bc -l: 计算器 (-l:浮点运算)
- # 为什么除以4,又除以1000000,计算的是million值
- echo "`zcat trt_N061011_1.fq.gz | wc-l` / (4*1000000)" | bc -l
- # 测序碱基数计算
- zcat trt_N061011_1.fq.gz | awk'{if(FNR%4==0) base =length}END{print base/10^9,"G";}'
2.基因组FASTA文件
此文件可以从ensemble数据库下载的(https://www.ensembl.org/info/data/ftp/index.html), 一般选择下载primary assemblyfasta(参考基因组和基因注释文件)。fasta文件用于序列存储,可以是DNA或蛋白序列,在此FASTA文件存储了基因组序列的信息。
序列名字行:以>符号开头,记录了该序列类型和所在基因组位置信息;
序列行(一行或多行):序列信息,soft-masked基因组会把所有重复区和低复杂区的序列用小写字母标出的基因组,小写字母n表示未知碱基。
- >1 dna_sm:chromosomechromosome:GRCh38:1:1:248956422:1 REF
- nnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnn
- nnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnn
- .....
- ttgggctggggcctggccatgtgtatttttttaaatttccactgatgattttgctgcatg
- gccggtgttgagaatgactgCGCAAATTTGCCGGATTTCCTTTGCTGTTCCTGCATGTAG
- TTTAAACGAGATTGCCAGCACCGGGTATCATTCACCATTTTTCTTTTCGTTAACTTGCCG
- .....
- nnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnnn
- # 通常要求序列名字行简单为好,而且一般加chr作为开头
- # 给第一行添加chr标签,并去掉其他多余信息
- # 下面的写法复杂了些,是为了避免给已经有chr信息的名字再加一次
- # 帮助无脑操作
- sed 's/^>([^chr])/>chr1/' Homo_sapiens.GRCh38.dna.primary_assembly.fa |cut -f 1 -d ' ' > GRCh38.fa
3. 基因组注释文件gff和gtf
gff全称General featureformat,主要是用来注释基因组。gtf全称Gene transfer format,主要是用来对基因进行注释。两者均是一个9列的基因信息注释文件,前8列的信息几乎一样,区别在于第9列。具体可见历史推文GTF/GFF文件格式解读和转换 在此不再赘述。
从ensemble下载的gtf文件前5行一般是以#开头的注释信息,后续分析中用不上需要去除,同时需要给第一列添加chr标签(与基因组序列一致),可通过下面的命令对文件进行加工:
- # grep 匹配查询 -v 输出不匹配的行
- gunzip Homo_sapiens.GRCh38.94.gtf.gz -c |grep -v '^#' | sed '/^[^chr]/ s/^/chr/' >GRCh38.gtf
4. bed文件
分析过程中的bed文件一般代表区域信息,如表示Peak位置的bed文件,表示基因注释的bed12文件。
- 表示基因注释时,gtf/gff和bed文件的区别
1)gtf/gff文件一行表示一个exon/CDS等子区域,多行联合表示一个gene;bed文件一行表示一个gene;
2)gtf文件中碱基位置定位方式是1-based,而bed中碱基定位方式是0-based,如下图所示。
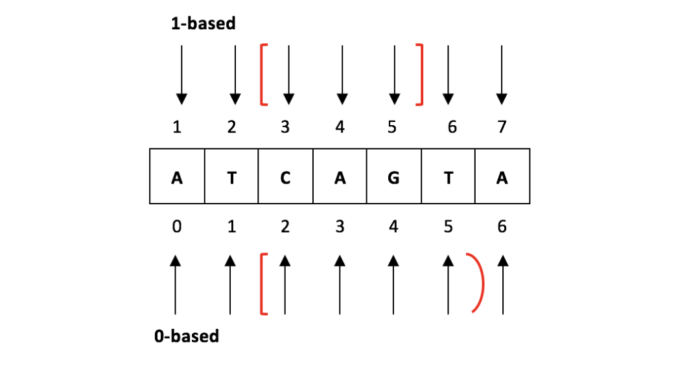
- bed文件每一行对应信息
必须包含的3列信息:
1)chrom:染色体名字 (e.g.chr3, chrY, chr2_random或者scaffold10671)。
2)chromStart:基因在染色体或scaffold上的起始位置(0-based)。
3)chromEnd:基因在染色体或scaffold上的终止位置 (前闭后开)。
可选的9列信息:
4)name:bed文件的行名。
5)score:本条基因在注释数据集文件中的评分(0-1000),在Genome Browser中会根据不同区段的评分显示对应的阴影强度(评分越高灰度越高)。
6)strand:链的方向 、-或. (.表示不确定链的方向)
7)thickStart:CDS区(编码区)的起始位置,即起始密码子的位置。
8)thickEnd:The endingposition at which the feature is drawn thickly (for example the stop codon ingene displays).
9)itemRgb:RGB颜色值(如:255,0,0),方便在GenomeBrowser中查看。
10)blockCount:bed行中外显子的数目。
11)blockSizes:逗号分割的列,数目与blockCount值对应,每个数表示对应外显子的碱基数。
12)blockStarts:逗号分割的列,数目与blockCount值对应,每个数表示对应外显子的起始位置(数值是相对ChromStart计算的)。
5. sam和bam文件
sam文件全称The SequencingAlignment/Map Format,是Alignment/Map步骤bwa/STAR/HISAT2等软件对结果的标准输出文件,用于存储reads比对到参考基因组的比对结果,是一个纯文本格式,文件一般较大。为了节省硬盘存储,一般使用其高效压缩的二进制格式bam文件。
利用samtools view的-b参数就能把sam文件转为bam文件。
1)sam文件查看方式
在linux终端直接用less即可进行查看;
2)bam文件查看方式
需要借助samtools view工具进行查看
- samtools view filename.bam | less -S
- samtools view -h filename.bam | less -S
NGS分析中大多数文件都是由header和record两部分组成,加上-h参数后可以将header显示出来,默认是不显示的。
- @HD VN:1.5 SO:coordinate
- @SQ SN:chr1 LN:248956422
- @SQ SN:chr10 LN:133797422
- ......
- @SQ SN:chrKI270392.1 LN:971
- @SQ SN:chrKI270394.1 LN:970
- @RG ID:BH_H3K27ac_2 LB:BH_H3K27ac_2 SM:BH_H3K27ac_2
- @PG ID:bwa PN:bwa VN:0.7.15-r1140 CL:bwa mem -M -t 8 -R@RGtID:BH_H3K27ac_2tLB:BH_H3K27ac_2tSM:BH_H3K27ac_2tPL: /MP
- @PG ID:MarkDuplicates VN:1.138(aa51703435dc6a423013e74e56b0b68405facd79_1439324166) CL:picard.sam.markduplicates.
- K00141:244:HVL3NBBXX:8:2119:27235:3145399 chr1 10016 32 115M = 10016 115 CCCTAACCCTAACCCTAACCC
- K00141:244:HVL3NBBXX:8:2119:27235:31453147 chr1 10016 32 115M = 10016 -115 CCCTTACCCTAACCCTAACCC
- header内容
@HD:是必须的标准文件头,包含版本信息;
@SQ:参考序列染色体名字和长度信息 (SN:scaffold name; LN: length);
@RG:重要read group信息,通常包含测序平台,测序文库和样本ID等信息,分析时用于区分不同样本(重测序时用到);
@PG:生成此文件的操作过程和参数信息 (program)。 - record内容
每一行就是一条read比对上参考基因组的信息,总共12列,用tab键分割。
- # 1. read名称;
- # 2. 比对信息位flag值;
- # 3. 参考序列染色体编号;
- # 4. 5′端起始位置;
- # 5. MAPQ:mapping quality,描述比对的质量,数字越大,特异性越高;
- # 6. CIGAR字符串,记录插入、删除、错配等信息;
- # 7. 配对read所比对到的染色体,仅双端测序的数据才有;
- # 8. 配对read所比对到的位置,仅双端测序的数据才有;
- # 9. 插入片段的长度,仅双端测序的数据才有;
- # 10. read序列;
- # 11. read质量值;
- # 12. 12列以后的信息都是metadata,程序用标记
sam文件中第二列flag信息很重要,下面做进一步解释。
利用samtools flagstat工具可以查看bam文件中比对的flag信息,并输出比对的统计结果。
- samtools flagstat *.bam
flag一共有12个标签,使用16进制数表示,每个标签值是2^(n-1),其中n<=12,每个值有其对应的唯一解释含义,具体见下图。
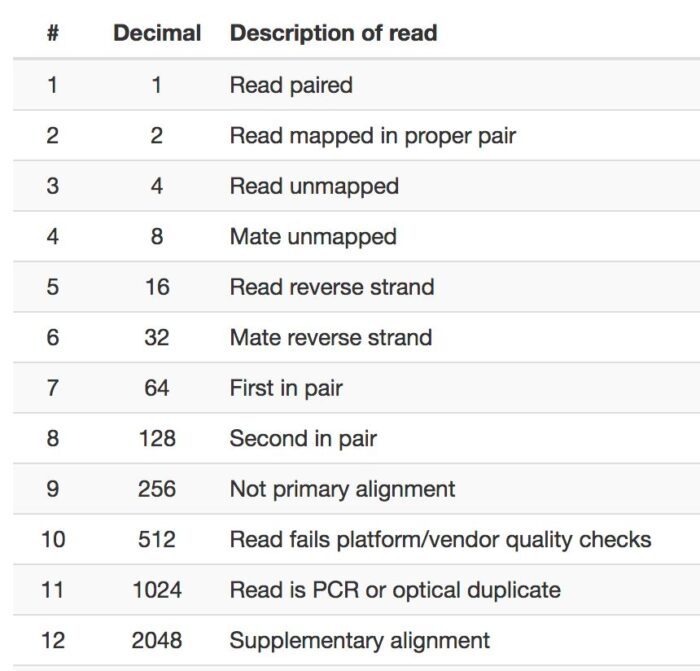
你会发现随机挑选几个值做加和运算,他们的结果都是唯一的,所以在bam文件中第二列flag的值代表这条序列符合下图所示条件的值的和。
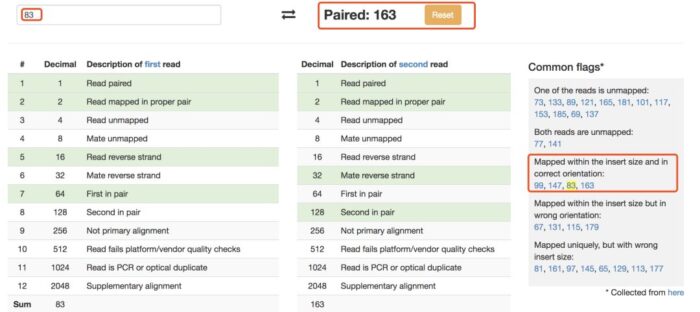
所以根据这个值我们可以判断这条序列是双端测序还是单端测序;如果是双端测序,reads来自左端还是右端。比如65 只能是1和64组成,代表这个序列是双端测序,而且是read1。
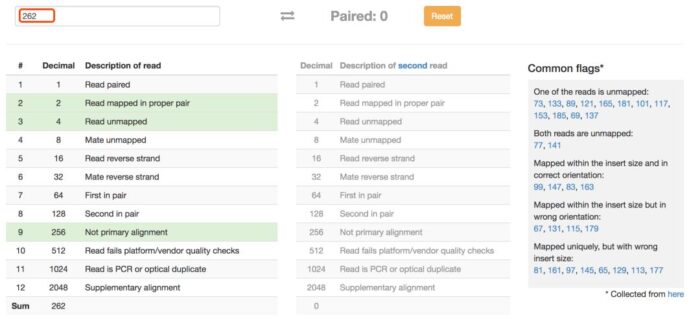
每次转换很头疼?别担心,网上有很多解码flag含义的在线工具,如SAM Format(网址:https://www.samformat.info/sam-format-flag)
输入flag的值,解析工具会返回匹配上的信息。如果是单端测序,flag值都是偶数。
如果是双端测序,工具会帮我们把另外一端序列的flag值返回,并且将这些数字情况大致分为5类,在右侧进一步显示这个值对应的含义。
6. wig、bigwig和bedgraph文件
上述bam和sam文件可以帮助我们探索reads在参考基因组中的比对情况,导入基因组浏览器查看比对状态和突变信息。而wiggle(简称wig)、bigwig(简写bw)以及bedgraph(简写bdg)只包含区域和区域的覆盖度信息,文件更小,用于可视化更方便,可以导入基因组浏览器(Genome Browser)中进行可视化,以查看reads在参考基因组各个区域的覆盖度并检测测序深度。这几个文件在ChIP-seq数据分析Call Peak阶段会生成,可以利用IGV等工具进行可视化,方便查看组蛋白修饰的程度。
wiggle:展示区域的密度或强度信息,如GCpercent,probability scores, andtranscriptome data.
- variableStep chrom=chr2
- 300701 12.5
- 300702 12.5
- 300703 12.5
- 300704 12.5
- 300705 12.5
- fixedStep chrom=chr3 start=400601 step=100span=5
- 11
- 22
- 33
bedGraph:bed与wig的结合,更省空间和灵活,展示信息与wig类似。(bedGraph的格式一般有四列,Chr、start、end和value,并且坐标是以0为起始左闭右开)
- chromA chromStartA chromEndA dataValueA
- chromB chromStartB chromEndB dataValueB
bigWig: wig文件的二进制压缩格式,可通过wigToBigWig工具转换
推荐大家阅读UCSC官网对这几个文件的详细解释:
- wiggle(WIG):https://genome.ucsc.edu/goldenPath/help/wiggle.html
- bedGraph:https://genome.ucsc.edu/goldenPath/help/bedgraph.html
- bigWig:http://genome.ucsc.edu/goldenPath/help/bigWig.html