

注:这里的cell是一语双关,既指表中的单元格,又指细胞。细胞就是单元格。
单细胞数据格式
为了记录每个细胞的信息,人们发展了相应的数据结构如seurat的S4类,monocle的CDS,SingleCellExperiment的sce,scanpy的anndata等,可见单细胞的故事远大于一张二维的表达谱。
seurat的S4类

- pbmc_small@commands
- $NormalizeData.RNA
- Command: NormalizeData(object = pbmc_small)
- Time: 2018-08-28 04:32:17
- assay : RNA
- normalization.method : LogNormalize
- scale.factor : 10000
- verbose : TRUE
- $RunPCA.RNA
- Command: RunPCA(object = pbmc_small, features = VariableFeatures(object = pbmc_small), verbose = FALSE)
- Time: 2018-08-28 04:34:56
- assay : RNA
- features : PPBP IGLL5 VDAC3 CD1C AKR1C3 PF4 MYL9 GNLY TREML1 CA2 SDPR PGRMC1 S100A8 TUBB1 HLA-DQA1 PARVB RUFY1 HLA-DPB1 RP11-290F20.3 S100A9
- compute.dims : 20
- rev.pca : FALSE
- weight.by.var : TRUE
- verbose : FALSE
- print.dims : 1 2 3 4 5
- features.print : 30
- reduction.name : pca
- reduction.key : PC
- seed.use : 42
- $BuildSNN.RNA.pca
- Command: BuildSNN(pbmc_small, features = VariableFeatures(object = pbmc_small))
- Time: 2018-08-28 04:43:31
- assay : RNA
- features : PPBP IGLL5 VDAC3 CD1C AKR1C3 PF4 MYL9 GNLY TREML1 CA2 SDPR PGRMC1 S100A8 TUBB1 HLA-DQA1 PARVB RUFY1 HLA-DPB1 RP11-290F20.3 S100A9
- reduction : pca
- dims : 1 2 3 4 5 6 7 8 9 10
- k.param : 30
- prune.SNN : 0.06666667
- nn.eps : 0
- verbose : TRUE
- force.recalc : FALSE
- do.plot : FALSE
- graph.name : RNA_snn
- ......
SingleCellExperiment
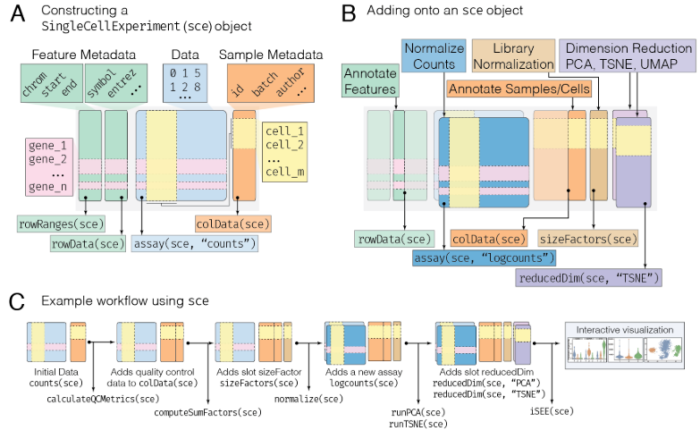
- A : 最小的sce对象是通过提供数据来构建的,比如每个细胞的计数矩阵(蓝色方框),由特征组成,比如基因(行)和细胞(列)。还可以提供描述单元格的元数据,其中单元格表示为行,单元格的已知特征为列(橙色框)。类似地,也可以添加描述特性的元数据(绿色框)。这些不同类型的数据都存储在sce对象的不同部分中,这些部分称为槽(slots)。每个槽中的数据可以通过以各自的槽(箭头)命名的访问器以编程方式访问,比如rowRanges指的是特征元数据,colData指的是样本元数据,assay指的是数据。
- B : 使用sce (singlecellexper, sce)兼容的工作流进行分析,将数据附加到初始sce对象。例如,计算每个单元格的库规范化因子将创建一个新槽(粉色框)。这些可以用来推导一个归一化计数矩阵,它与初始计数数据(深蓝色方框)一起存储在同一个检测槽中。因此,分析槽能够存储任意数量的数据转换。单元质量度量(描述单元特征)被附加到样例元数据槽colData中。最后,以与分析槽类似的方式,可以存储任意数量的维数缩减的数据表示形式,驻留在它们自己的槽中,reducedDim。
- C : sce对象在典型分析的整个过程中不断发展,存储来自初始数据的各种度量和表示。有关singlecellexper类的更多信息,请参见singlecellexper(https://bioconductor.org/packages/singlecellexper)。
anndata
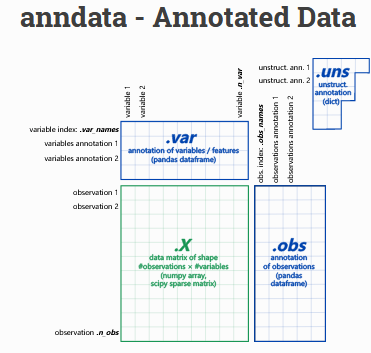
单细胞转录组的核心就是一个cell X gene的二维表,但是分群后要存储cell的分群结果,特征选择是要记录gene的信息,降维后要存储降维后的结果。所以,这张表.X的对象cell相关的信息记录在.obs中,属性gene的信息记录在.var中,其他的信息在.uns中。
记得初中时学习立体几何老师要求我们要有空间想象力,把思维提高到一个新的维度。在单细胞数据分析的过中,我们也要挑起我们的想象力,比如在RNA速率的分析中,anndata存储的内容是这样的:
- adata
- AnnData object with n_obs × n_vars = 7292 × 1999
- obs: 'initial_size_unspliced', 'initial_size_spliced', 'initial_size', 'n_counts', 'velocity_self_transition', 'leiden', 'velocity_clusters'
- var: 'Accession', 'Chromosome', 'End', 'Start', 'Strand', 'means', 'dispersions', 'dispersions_norm', 'velocity_gamma', 'velocity_r2', 'velocity_genes', 'velocity_score', 'fit_alpha', 'fit_beta', 'fit_gamma', 'fit_t_', 'fit_scaling', 'fit_std_u', 'fit_std_s', 'fit_likelihood', 'fit_u0', 'fit_s0', 'fit_pval_steady', 'fit_steady_u', 'fit_steady_s', 'fit_alignment_scaling', 'fit_r2'
- uns: 'pca', 'neighbors', 'connectivities_key', 'distances_key', 'velocity_settings', 'velocity_graph', 'velocity_graph_neg', 'leiden', 'umap', 'leiden_colors', 'rank_velocity_genes', 'recover_dynamics'
- obsm: 'X_pca', 'X_umap', 'velocity_umap'
- varm: 'PCs', 'loss'
- layers: 'matrix', 'ambiguous', 'spliced', 'unspliced', 'Ms', 'Mu', 'velocity', 'variance_velocity', 'fit_t', 'fit_tau', 'fit_tau_', 'velocity_u'
- obsp: 'distances', 'connectivities'
我们不仅要知道每一部分存储的内容,还要知道各部分之间的关系。
h5
h5文件是层次格式的第5代版本,用于存储科学数据的一种文件格式和库文件,由美国超级计算中心与应用中心研发的文件格式,用以存储和组织大规模数据.
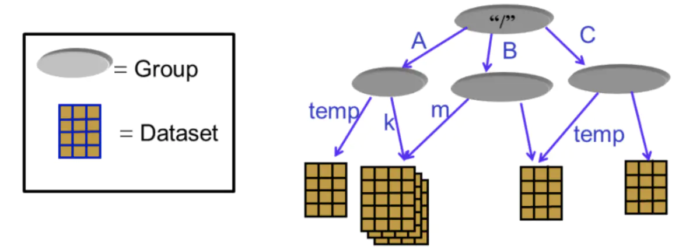
H5将文件结构简化成两个主要的对象类型:
1 数据集dataset,就是同一类型数据的多维数组
2 组group,是一种容器结构,可以包含数据集和其他组,若一个文件中存放了不同种类的数据集,这些数据集的管理就用到了group
直观的理解,可以参考我们的文件系统,不同的文件存放在不同的目录下:
目录就是hdf5文件中的group,描述了数据集DataSet的分类信息,通过group有效的将多种dataset进行管理和划分文件就是hdf5文件中的dataset,表示具体的数据
下图就是数据集和组的关系:
为什么是数据库
随着单细胞多模态数据的丰富,目前只是转录组的数据结构就这么丰富了,想想一下每个细胞还有表观信息,以后还有空间信息,多组学的发展一定会再次丰富数据结构的。
另一个层面的是,数据结构是和算法结合在一起的。算法就是处理某一问题的解法,过程和步骤,不同的数据结构会简化算法复杂度。数据结构是描述问题,算法是解决问题,二者是紧密联系在一起的。
而数据库的概念把我们从数据表的狭小空间中解放出来:数据库系统是为适应数据处理的需要而发展起来的一种较为理想的数据处理系统,也是一个为实际可运行的存储、维护和应用系统提供数据的软件系统,是存储介质 、处理对象和管理系统的集合体。
其实数据库在单细胞数据分析中已经很常见了,除了上面我们提到的常见的数据格式之外,有不少研究者会把数据放到公用数据库中。只是这类的大数据库,初学者会觉得离自己很遥远。
数据库之外
我们花大力气去存储数据,描述数据,是为了什么呢?或者回到问题的开始,就是一张二维表,我们能得到什么信息呢?所以很多时候热闹是他们的,我们的关注点还是应该放在生物学的问题上。以技术为舟帆去游弋生物学的海洋。