LIGER简介
LIGER能够跨个体、物种和方法(基因表达、表观遗传或空间数据)识别共有的细胞类型,以及数据集特有的特征,提供对不同单细胞数据集的统一分析。
LIGER原理
接触生信的朋友对PCA都有一定了解,它可以用一个低维矩阵(行比较少的矩阵)表示高维矩阵(行比较多的矩阵)的大部分信息。类似的降维方法还有一些,比如奇异值分解(SVD)、非负矩阵分解(NMD)等,liger所用的方法是NMD的改良版——综合非负矩阵分解(iNMD)。具体的算法实现咱们不必苛求理解掌握,知道以下几点即可:
单细胞基因表达矩阵经过iNMD分解后,可以得到三个矩阵:因子(行) x 细胞 (列)的H矩阵,数据集特有基因(行) x 因子 (列)的V矩阵,以及数据集共有基因(行) x 因子 (列)的W矩阵。 iNMD分解矩阵后的“因子”,相当于PCA降维之后的"PC"轴;但是iNMD的因子比 PCA的PC轴可解释性更强,在单细胞数据分析中,每个因子常常对应一种细胞类型。 V矩阵和W矩阵中loading值靠前的基因,往往具有显著的生物学意义。
原文图解及说明如下:
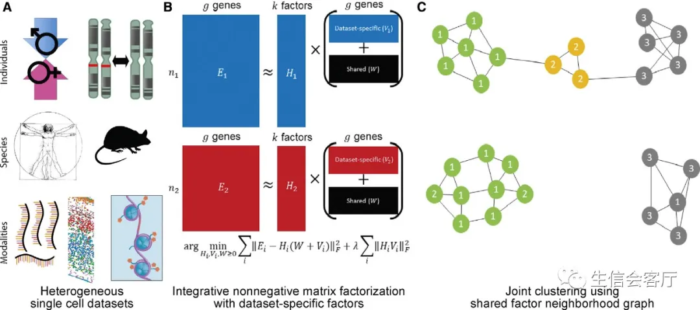
(A) LIGER takes as input two or more datasets, which may come from different individuals, species, or modalities, that share corresponding gene-level features. (B) Integrative nonnegative matrix factorization (Yang and Michailidis, 2016) identifies shared and dataset-specific metagenes across datasets. (C) Building a graph in the resulting factor space, based on comparing neighborhoods of maximum factor loadings (STAR Methods). Each cell is numbered by its maximum factor loading and connected to its nearest neighbors within each dataset. The shared factor neighborhood graph leverages the factor loading values of neighboring cells to prevent the spurious integration of divergent cell types across datasets (such as the yellow cells shown).
安装LIGER包
除了系统要有java之外,没有特别依赖的库。很容易安装的一个包,装不上多半是网络问题。
- library(devtools)
- install_github('MacoskoLab/liger')
scRNA多数据集的整合
为了方便与seurat和harmony的效果对比,继续使用《单细胞转录组分析:多样本合并与批次校正》一文中使用的那10个样本的数据。
- ##==准备seurat对象列表==##
- library(Seurat)
- library(tidyverse)
- rm(list=ls())
- setwd("~/project/harmony")
- dir <- dir("GSE139324/") #GSE139324是存放数据的目录
- dir <- paste0("GSE139324/",dir)
- sample_name <- c('HNC01PBMC', 'HNC01TIL', 'HNC10PBMC', 'HNC10TIL', 'HNC20PBMC',
- 'HNC20TIL', 'PBMC1', 'PBMC2', 'Tonsil1', 'Tonsil2')
- scRNAlist <- list()
- for(i in 1:length(dir)){
- counts <- Read10X(data.dir = dir[i])
- scRNAlist[[i]] <- CreateSeuratObject(counts, project=sample_name[i], min.cells=3, min.features = 200)
- scRNAlist[[i]][["percent.mt"]] <- PercentageFeatureSet(scRNAlist[[i]], pattern = "^MT-")
- scRNAlist[[i]] <- subset(scRNAlist[[i]], subset = percent.mt < 10)
- }
- saveRDS(scRNAlist, "scRNAlist.rds")
LIGER整合数据
- ##==多个scRNA-seq数据整合==##
- library(Seurat)
- library(liger)
- library(tidyverse)
- library(patchwork)
- library(SeuratWrappers)
- #为了方便展示效果,只取其中的2个样本演示
- scRNA <- merge(scRNAlist[[2]], scRNAlist[[10]]) -> scRNA.orig
- scRNA <- NormalizeData(scRNA)
- scRNA <- FindVariableFeatures(scRNA)
- scRNA <- ScaleData(scRNA, split.by="orig.ident", do.center=FALSE)
- nFactors=20 #设置矩阵分解的因子数,一般取值20-40
- ##因式分解
- scRNA <- RunOptimizeALS(scRNA, k=nFactors, split.by="orig.ident")
- ##多样本整合
- scRNA <- RunQuantileNorm(scRNA, split.by="orig.ident")
- #整理因子顺序
- scRNA$clusters <- factor(scRNA$clusters,
- levels=1:length(levels(scRNA$clusters)))
- ##聚类
- scRNA <- FindNeighbors(scRNA, reduction="iNMF",
- dims=1:nFactors) %>% FindClusters()
- scRNA <- RunUMAP(scRNA, dims=1:nFactors, reduction="iNMF")
- ##不整合数据的降维聚类
- scRNA.orig <- scRNA.orig %>% NormalizeData() %>% FindVariableFeatures() %>% ScaleData() %>%
- RunPCA() %>% FindNeighbors(dims=1:20) %>% FindClusters() %>% RunUMAP(dims=1:20)
- ##可视化
- p1 = DimPlot(scRNA, group.by="orig.ident", pt.size=0.05) ggtitle("Integrated by liger")
- p2 = DimPlot(scRNA.orig, group.by="orig.ident", pt.size=0.05) ggtitle("No integrated")
- p3 = DimPlot(scRNA, group.by="seurat_clusters", label=T, label.size=2) ggtitle("Clustered by seurat")
- p4 = DimPlot(scRNA, group.by="clusters", label=T, label.size=2) ggtitle("Clustered by liger")
- plot1 = p1 p2 plot_layout(guides = 'collect')
- plot2 = p3|p4
- ggsave("integrated_liger.png", plot=plot1, width=8, height=3.6)
- ggsave("clustered_liger.png", plot=plot2, width=8, height=3.6)
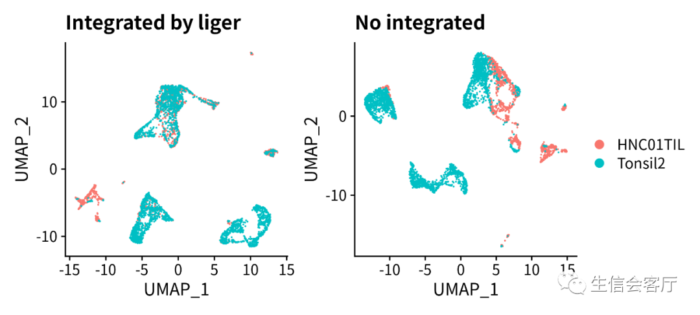
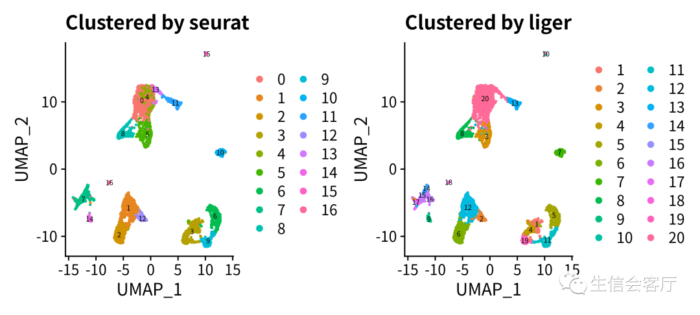
经过liger整合的数据,meta.data中会有两个聚类的结果。seurat_clusters列是seurat聚类的结果,clusters列是liger聚类的结果,其聚类数量与RunOptimizeALS函数运行时k参数的值相同。
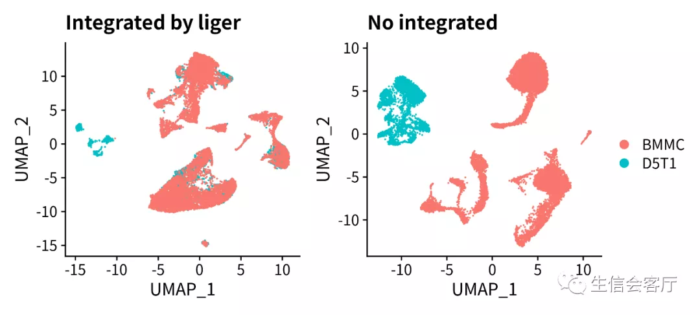
scRNA与scATAC的整合
这部分使用liger教程提供的示例数据,下载地址https://umich.box.com/s/wip2nzpktn6fdnlpc83o1u7anjn4ue2c
- ##===scRNA与scATAC的整合===##
- ##数据准备
- scATAC <- readRDS('data/GSM4138888_scATAC_BMMC_D5T1.RDS')
- scRNA1 <- readRDS('data/GSM4138872_scRNA_BMMC_D1T1.rds')
- scRNA2 <- readRDS('data/GSM4138873_scRNA_BMMC_D1T2.rds')
- scATAC <- aggregate(scATAC, by=list(as.factor(rownames(scATAC))), FUN=sum)
- tmp <- scATAC$Group.1
- scATAC <- as.matrix(scATAC[,-1])
- rownames(scATAC) <- tmp
- scATAC <- CreateSeuratObject(scATAC, min.cells=3, min.features = 200)
- scRNA1 <- CreateSeuratObject(scRNA1, min.cells=3, min.features = 200)
- scRNA2 <- CreateSeuratObject(scRNA2, min.cells=3, min.features = 200)
- #合并后的数据生成两个副本
- scRNA <- merge(scATAC, list(scRNA1, scRNA2)) -> scRNA.orig
- ##liger整合流程
- #数据标准化
- scRNA <- NormalizeData(scRNA)
- scRNA <- FindVariableFeatures(scRNA)
- scRNA <- ScaleData(scRNA, split.by="orig.ident", do.center=FALSE)
- nFactors=20 #设置矩阵分解的因子数,一般取值20-40
- #因式分解
- scRNA <- RunOptimizeALS(scRNA, k=nFactors, split.by="orig.ident")
- #多样本整合
- scRNA <- RunQuantileNorm(scRNA, split.by="orig.ident")
- #整理因子顺序
- cRNA$clusters <- factor(scRNA$clusters, levels=1:length(levels(scRNA$clusters)))
- #聚类
- scRNA <- FindNeighbors(scRNA, reduction="iNMF", dims=1:nFactors) %>% FindClusters()
- scRNA <- RunUMAP(scRNA, dims=1:nFactors, reduction="iNMF")
- ##不整合数据的降维聚类
- scRNA.orig <- scRNA.orig %>% NormalizeData() %>% FindVariableFeatures() %>% ScaleData() %>%
- RunPCA() %>% FindNeighbors(dims=1:20) %>% FindClusters() %>% RunUMAP(dims=1:20)
- ##可视化
- p1 = DimPlot(scRNA, group.by="orig.ident", pt.size=0.05) ggtitle("Integrated by liger")
- p2 = DimPlot(scRNA.orig, group.by="orig.ident", pt.size=0.05) ggtitle("No integrated")
- p3 = DimPlot(scRNA, group.by="seurat_clusters", label=T, label.size=3) ggtitle("Clustered by seurat")
- p4 = DimPlot(scRNA, group.by="clusters", label=T, label.size=3) ggtitle("Clustered by liger")
- plot1 = p1 p2 plot_layout(guides = 'collect')
- plot2 = p3|p4
- ggsave("atac-rna_integrated_liger.png", plot=plot1, width=8, height=3.6)
- ggsave("atac-rna_clustered_liger.png", plot=plot2, width=8, height=3.6)
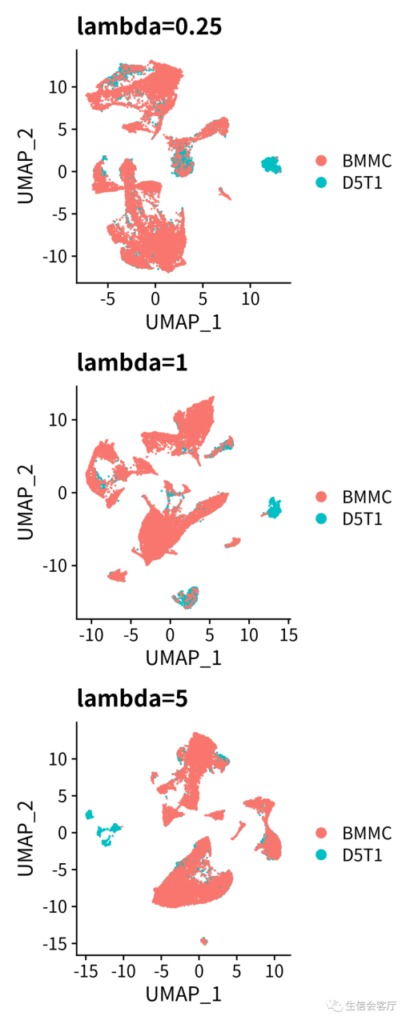
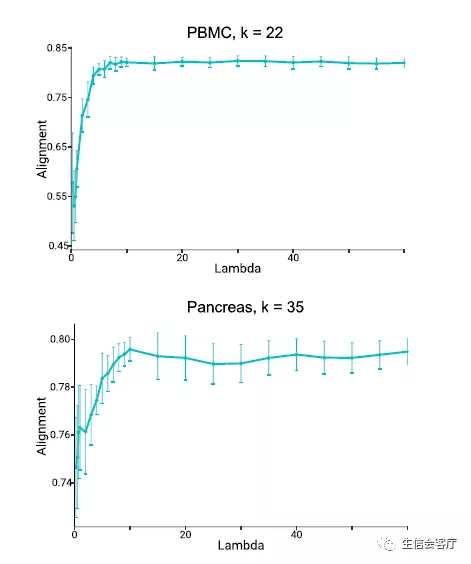
调整整合力度
文献中提到liger可以通过lambda参数调整对齐程度(alignment),原文用了两个数据集进行测试,lambda参数对alignment的影响如下图所示:
我也测试了一下直观效果,代码如下:
- ##===测试lambda===##
- scRNA.x <- merge(scATAC, list(scRNA1, scRNA2)) -> scRNA.y
- ##scRNA.x测试lambda=0.25
- scRNA.x <- NormalizeData(scRNA.x)
- scRNA.x <- FindVariableFeatures(scRNA.x)
- scRNA.x <- ScaleData(scRNA.x, split.by="orig.ident", do.center=FALSE)
- nFactors=20 #设置矩阵分解的因子数,一般取值20-40
- #因式分解
- scRNA.x <- RunOptimizeALS(scRNA.x, k=nFactors, lambda=0.25, split.by="orig.ident")
- #多样本整合
- scRNA.x <- RunQuantileNorm(scRNA.x, split.by="orig.ident")
- #整理因子顺序
- scRNA.x$clusters <- factor(scRNA.x$clusters, levels=1:length(levels(scRNA.x$clusters)))
- #聚类
- scRNA.x <- FindNeighbors(scRNA.x, reduction="iNMF", dims=1:nFactors) %>% FindClusters()
- scRNA.x <- RunUMAP(scRNA.x, dims=1:nFactors, reduction="iNMF")
- ##scRNA.y测试lambda=1
- scRNA.y <- NormalizeData(scRNA.y)
- scRNA.y <- FindVariableFeatures(scRNA.y)
- scRNA.y <- ScaleData(scRNA.y, split.by="orig.ident", do.center=FALSE)
- nFactors=20 #设置矩阵分解的因子数,一般取值20-40
- #因式分解
- scRNA.y <- RunOptimizeALS(scRNA.y, k=nFactors, lambda=1, split.by="orig.ident")
- #多样本整合
- scRNA.y <- RunQuantileNorm(scRNA.y, split.by="orig.ident")
- #整理因子顺序
- scRNA.y$clusters <- factor(scRNA.y$clusters, levels=1:length(levels(scRNA.y$clusters)))
- #聚类
- scRNA.y <- FindNeighbors(scRNA.y, reduction="iNMF", dims=1:nFactors) %>% FindClusters()
- scRNA.y <- RunUMAP(scRNA.y, dims=1:nFactors, reduction="iNMF")
- p1 = DimPlot(scRNA, group.by="orig.ident", pt.size=0.05) ggtitle("lambda=5")
- p2 = DimPlot(scRNA.x, group.by="orig.ident", pt.size=0.05) ggtitle("lambda=0.25")
- p3 = DimPlot(scRNA.y, group.by="orig.ident", pt.size=0.05) ggtitle("lambda=1")
- p = p2/p3/p1
- ggsave("lambda_test.png", plot=p, width=4, height=10)