

速率分析的原理
大家第一次听到RNA速率/速度,是不是感觉和我一样懵呢?再看文献提到的时间导数、微分方程和一大堆公式,是不是更搞不清作者在讲什么?我们常用的基因表达矩阵,如velocyto的作者在文献中所讲,反映的是细胞转录组的瞬间快照。其实在生命体中,每时每刻都发生着mRNA的转录、剪接和降解,它们当然都是有速度的,作者在文中用α、β和γ表示这些速度。不仅mRNA的转录、剪接和降解有速度,前体mRNA和成熟mRNA的丰度变化也有速度,作者所讲的RNA速度特指成熟mRNA的丰度变化速度。RNA速度可以通过前体mRNA和成熟mRNA的指标来估计,并且可以用来预测细胞未来数小时内的状态。在velocyto的动力学模型中,作者假定转录速度 α恒定,此时unspliced mRNA(u)和未来的spliced mRNA(s)的丰度高度相关:
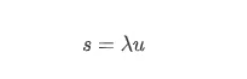
系数 λ 综合了降解率和剪接率、基因调控特性、内含子和外显子长度的比值以及内部启动位点的数量等因素。绝大多数的基因在不同细胞类型中,系数 λ 固定不变,但是11%的基因在不同组织中 λ 值不同。为了证明此模型的正确性,作者使用小鼠肝脏的节律基因来验证:
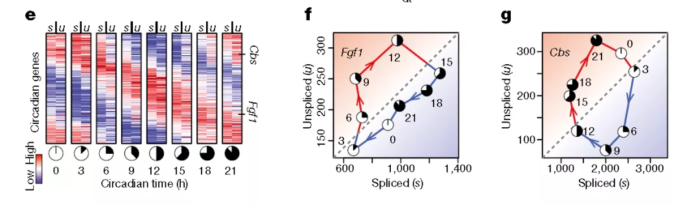
总的来说:unspliced mRNA丰度与spliced mRNA丰度高度相关且具有先导性,因此可以据此推断细胞的分化方向与速率。
velocyto分析流程
使用python版velocyto将bam文件转换为包含spliced,unspliced和ambiguous三个矩阵的loom文件; 读取 loom文件并转换为surat对象; 对seurat对象执行数据标准化及降维聚类操作; 计算RNA速率; 展示细胞分化的方向。
velocyto分析示例
本文演示数据来自velocyto官方教程:http://pklab.med.harvard.edu/velocyto/notebooks/R/SCG71.nb.html
- library(Seurat)
- library(velocyto.R)
- library(tidyverse)
- library(SeuratWrappers)
- ##数据基础分析
- # 读取loom文件
- velo <- read.loom.matrices(url("http://pklab.med.harvard.edu/velocyto/mouseBM/SCG71.loom"))
- # 转换为seurat对象
- velo <- as.Seurat(x = velo)
- # 降维聚类
- velo <- velo %>% SCTransform(assay="spliced") %>% RunPCA(verbose=F)
- ElbowPlot(velo, ndims = 50)
- nPC=1:20
- velo <- FindNeighbors(velo, dims = nPC) %>% FindClusters() %>%
- RunUMAP(dims = nPC) %>% RunTSNE(dims = nPC)
- ##给细胞分配颜色
- ident.colors <- (scales::hue_pal())(n = length(x = levels(x = velo)))
- names(x = ident.colors) <- levels(x = velo)
- cell.colors <- ident.colors[Idents(object = velo)]
- names(x = cell.colors) <- colnames(x = velo)
- ##速率分析
- velo <- RunVelocity(velo, deltaT = 1, kCells = 25, fit.quantile = 0.02,
- spliced.average = 0.2, unspliced.average = 0.05, ncores = 18)
- #kCells:用于斜率平滑度计算最近邻细胞数量,越大越平滑,越小越能反映局部特征
- #fit.quantile:0.02代表对基因表达量最高2%与最低2%的值执行gamma拟合
- #spliced.average:过滤低表达丰度基因的标准,计算的是基因在cluster内的平均counts值
- #unspliced.average:同上
- ##全局速率可视化
- emb = Embeddings(velo, reduction = "umap")
- vel = Tool(velo, slot = "RunVelocity")
- show.velocity.on.embedding.cor(emb = emb, vel = vel, n = 200, scale = "sqrt",
- cell.colors = ac(cell.colors, alpha = 0.5), cex = 0.8, arrow.scale = 3,
- show.grid.flow = TRUE, min.grid.cell.mass = 0.5, grid.n = 40,
- arrow.lwd = 1, do.par = FALSE, cell.border.alpha = 0.1)
- ##特定基因速率可视化
- gene = "Camp"
- RunVelocity(velo, deltaT=1, kCells=25, fit.quantile=0.02, old.fit=vel,
- cell.emb=emb, cell.colors=cell.colors, show.gene=gene, do.par=T)
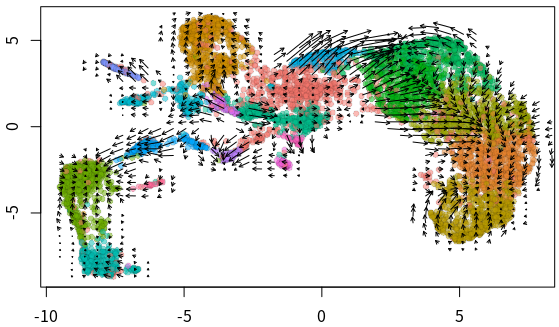
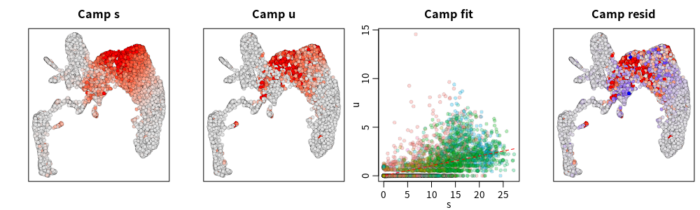
velocyto分析难点
bam转loom:bam文件转loom矩阵文件需要使用python版的velocyto,安装使用都比较困难,经常会有意想不到的报错。我可以提供有偿服务,帮大家完成bam文件的转换处理。 velocyto.R安装:R语言版的velocyto.R安装比较复杂,需要安装几个Linux库文件,初学者很难自己完成。 Seurat与velocyto整合:很多人希望从seurat的结果中抽取一个亚群来做速率分析,但是velocyto从bam文件得到表达矩阵是所有细胞的,降维聚类的结果与seurat也有差异,因此初学者很难将两者整合在一起。