Garnett简介
分析原理
Garnett使用人工定义的marker基因信息来选择细胞,然后基于这些细胞使用弹性网络回归(elastic-net regression)的机器学习算法训练分类器。分类器中定义细胞类型的基因不仅有marker file中人工挑选的marker,还会增加一些机器学习得到的新marker,利用这些信息就可以对新数据集的细胞分类。
分析流程
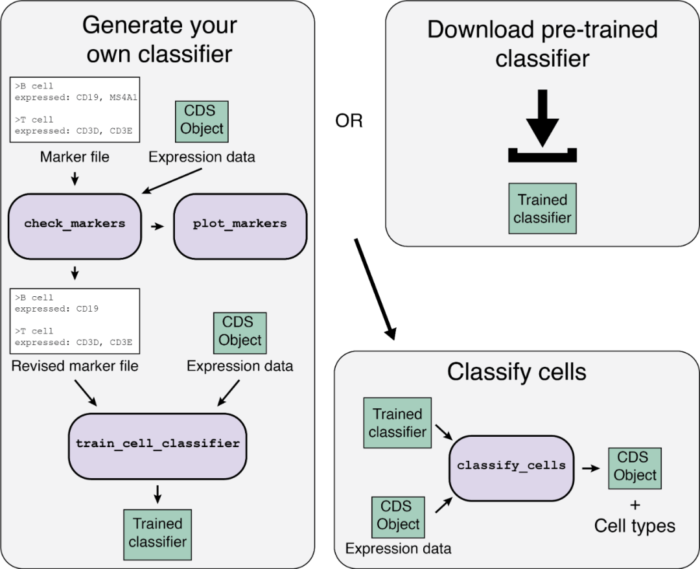
用户可以自己定义细胞类型的marker基因并训练分类器,也可以下载作者构建好的分类。
链接:https://cole-trapnell-lab.github.io/garnett/classifiers/
如果自己训练分类器,流程如下:
制作符合garnett格式要求的定义细胞类型的marker基因文本文件(marker file)。 使用单细胞数据创建monocle3的CDS数据对象(cds object)。 将marker file和cds object输入garnett,对marker基因进行打分,然后根据评分结果优化marker file。 将优化后的marker file和cds object输入garnett训练分类器。 使用分类器对新的cds数据进行细胞分类。
分类效果
Garnett是一种有监督的分类方法,其准确性和人工定义的marker基因信息密切相关。作者构建了几个分类器并测试了分类准确性,其中PBMC的结果最好:使用扩展分类方法,可以正确识别94%的细胞,剩下的2%分类错误,还有4%的细胞garnett无法识别。
安装garnett
Garnett的运行依赖monole3和bioconductor的物种基因信息数据库(org.xx.eg.db系列),安装garnett要先安装monole3和其他依赖包。
- ## 首先安装monole3
- # 安装monole3的依赖包
- BiocManager::install(c('BiocGenerics', 'DelayedArray', 'DelayedMatrixStats',
- 'limma', 'S4Vectors', 'SingleCellExperiment',
- 'SummarizedExperiment'))
- # 安装monocle3
- devtools::install_github('cole-trapnell-lab/monocle3')
- ## 然后安装常用的人和小鼠的基因信息数据库
- BiocManager::install(c('org.Hs.eg.db', 'org.Mm.eg.db'))
- ## 最后安装garnett
- devtools::install_github("cole-trapnell-lab/garnett", ref="monocle3")
10x PBMC数据实测
测试数据来源于10x genomics官网的示例数据集。
数据下载
- 下载PBMC单细胞数据,marker file和预先训练好的分类器。
- library(Seurat)
- library(monocle3)
- library(garnett)
- library(SingleR)
- library(org.Hs.eg.db)
- library(tidyverse)
- library(patchwork)
- rm(list=ls())
- dir.create("garnett_demo")
- setwd("garnett_demo")
- download.file(url="https://cf.10xgenomics.com/samples/cell-exp/3.0.2/5k_pbmc_v3_nextgem/5k_pbmc_v3_nextgem_filtered_feature_bc_matrix.h5",
- destfile = "pbmc.h5")
- download.file(url="https://cole-trapnell-lab.github.io/garnett/classifiers/hsPBMC_20191017.RDS",
- destfile = "hsPBMC.rds")
- download.file(url="https://cole-trapnell-lab.github.io/garnett/marker_files/hsPBMC_markers.txt",
- destfile = "hsPBMC_markers.txt")
- ## 创建seurat对象并降维聚类
- pbmc <- Read10X_h5("pbmc.h5")
- pbmc <- CreateSeuratObject(pbmc, min.cells = 3, min.features = 200)
- pbmc <- SCTransform(pbmc)
- pbmc <- RunPCA(pbmc, verbose = F)
- ElbowPlot(pbmc)
- pc.num=1:15
- pbmc <- pbmc %>% RunTSNE(dims=pc.num) %>% RunUMAP(dims=pc.num) %>%
- FindNeighbors(dims = pc.num) %>% FindClusters(resolution=0.8)
MarkerFile格式
训练分类器首先需要确定各种细胞类型的marker基因,并创建一个符合garnett要求的数据文件。以我们下载的marker file为例:

每个细胞类型必须包含两行:
“>”开头的细胞类型字段; “ expressed:” 开头的字符定义细胞类型的marker基因。
可选以下几行附加信息:“ not expressed:” 开头的字符定义细胞类型的负选marker基因。可以使用“ expressed above:”、“expressed below:”、“expressed between:”定义marker基因的表达值范围。“ subtype of:” 开头的字符定义细胞类型的父类,即此类细胞属于哪种细胞的亚型。“ references:” 开头的字符定义marker基因的选择依据。
Marker基因评估
- ## 创建CDS对象
- data <- GetAssayData(pbmc, assay = 'RNA', slot = 'counts')
- cell_metadata <- pbmc@meta.data
- gene_annotation <- data.frame(gene_short_name = rownames(data))
- rownames(gene_annotation) <- rownames(data)
- cds <- new_cell_data_set(data,
- cell_metadata = cell_metadata,
- gene_metadata = gene_annotation)
- #preprocess_cds函数相当于seurat中NormalizeData ScaleData RunPCA
- cds <- preprocess_cds(cds, num_dim = 30)
- ## 演示利用marker file训练分类器
- # 对marker file中marker基因评分
- marker_check <- check_markers(cds, "hsPBMC_markers.txt",
- db=org.Hs.eg.db,
- cds_gene_id_type = "SYMBOL",
- marker_file_gene_id_type = "SYMBOL")
- plot_markers(marker_check)
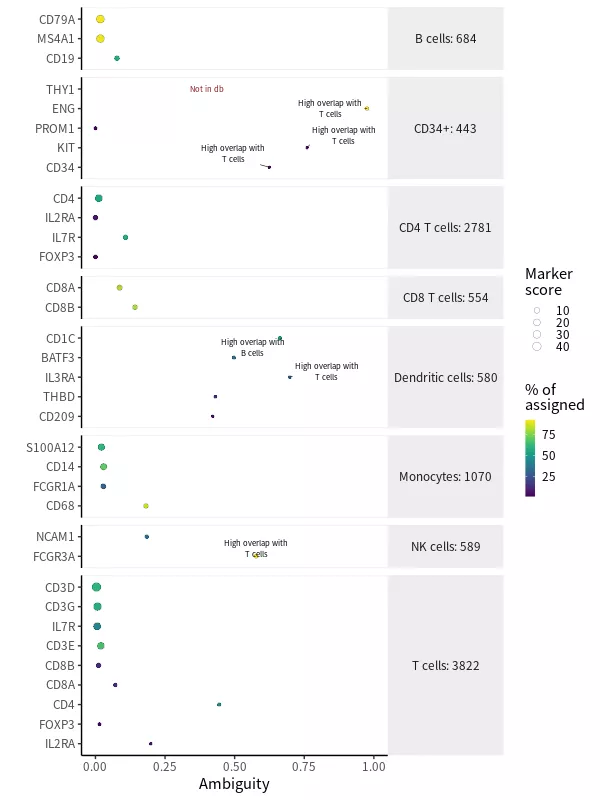
评估结果会以红色字体提示哪些marker基因在数据库中找不到对应的Ensembl名称,以及哪些基因的特异性不高(标注“High overlap with XX cells”)。我们可以根据评估结果优化marker基因,或者添加其他信息来辅助区分细胞类型。
训练分类器
- # 使用marker file和cds对象训练分类器
- pbmc_classifier <- train_cell_classifier(cds = cds,
- marker_file = "hsPBMC_markers.txt",
- db=org.Hs.eg.db,
- cds_gene_id_type = "SYMBOL",
- num_unknown = 50,
- marker_file_gene_id_type = "SYMBOL")
- saveRDS(pbmc_classifier, "pbmc_classifier.rds")
- # 查看分类器最后选择的根节点基因,注意markerfile的基因都会在其中
- feature_genes_root <- get_feature_genes(pbmc_classifier, node = "root",
- db = org.Hs.eg.db, convert_ids = TRUE)
- head(feature_genes_root)
- # B cells CD34 Dendritic cells Monocytes NK cells T cells Unknown
- #(Intercept) -0.17904570 -0.511471201 -0.46133695 -0.18514496 0.15949539 0.34180682 0.835696611
- #MS4A1 0.62437483 0.014971998 -0.51071341 -0.39488804 -0.04377010 -0.06051123 0.370535955
- #CD3E -0.04250250 0.113640425 -0.06674712 -0.14783769 -0.04573743 0.18596204 0.003222269
- #CD3D -0.21560305 -0.095353417 0.18417484 -0.03694182 -0.20621575 0.23510820 0.134830996
- #CD3G -0.07838514 0.003340245 0.04782908 -0.23665376 -0.01593186 0.21938815 0.060413288
- #CD19 1.05377553 -0.395188459 0.10976294 0.02730268 -0.10411129 -0.79719777 0.105656378
- # 查看分类器中分支节点的分类基因
- feature_genes_branch <- get_feature_genes(pbmc_classifier, node = "T cells",
- db = org.Hs.eg.db, convert_ids = TRUE)
- #head(feature_genes_branch)
- # CD4 T cells CD8 T cells Unknown
- #(Intercept) 0.776393262 -1.070693475 0.294300213
- #AKR7A2 0.009892929 0.003568803 -0.013461732
- #YTHDF2 -0.009176618 0.002927022 0.006249596
- #AGO3 -0.018056956 0.003335809 0.014721147
- #SCP2 0.066925112 0.024191726 -0.091116838
- #CDC14A 0.049606392 -0.006880622 -0.042725770
使用分类器
我们使用garnett官网训练好的分类器预测咱们下载的pbmc数据。
- ## 使用下载的分类器分类
- hsPBMC <- readRDS("hsPBMC.rds")
- pData(cds)$garnett_cluster <- pData(cds)$seurat_clusters
- cds <- classify_cells(cds, hsPBMC, db = org.Hs.eg.db, cluster_extend = TRUE, cds_gene_id_type = "SYMBOL")
- ## 将结果返回给seurat对象# 提取分类结果
- cds.meta <- subset(pData(cds), select = c("cell_type", "cluster_ext_type")) %>% as.data.frame()
- pbmc <- AddMetaData(pbmc, metadata = cds.meta)
分类结果对比
Azimuth分类
Azimuth的详细说明和使用方法见《单细胞分析之monocle3》中的利用azimuth鉴定细胞类型部分。
- ## 细胞分类之Azimuth
- pbmc_counts <- pbmc@assays$RNA@counts
- saveRDS(pbmc_counts, "pbmc_counts.rds")
- #上传http://azimuth.satijalab.org/app/azimuth网站在线分类,分类结果为azimuth_pred.tsv文件
- predictions <- read.delim('azimuth_pred.tsv', row.names = 1)
- pbmc <- AddMetaData(pbmc, metadata = predictions)
SingleR分类
- ## 细胞分类之SingleR
- refdata <- HumanPrimaryCellAtlasData()
- testdata <- GetAssayData(pbmc, slot="data")
- clusters <- pbmc$seurat_clusters
- cellpred <- SingleR(test = testdata, ref = refdata, labels = refdata$label.main,
- method = "cluster", clusters = clusters,
- assay.type.test = "logcounts", assay.type.ref = "logcounts")
- celltype = data.frame(ClusterID=rownames(cellpred), celltype=cellpred$labels, stringsAsFactors = F)
- pbmc$SingleR = "NA"
- for(i in 1:nrow(celltype)){
- pbmc$SingleR[which(pbmc$seurat_clusters == celltype$ClusterID[i])] <- celltype$celltype[i]}
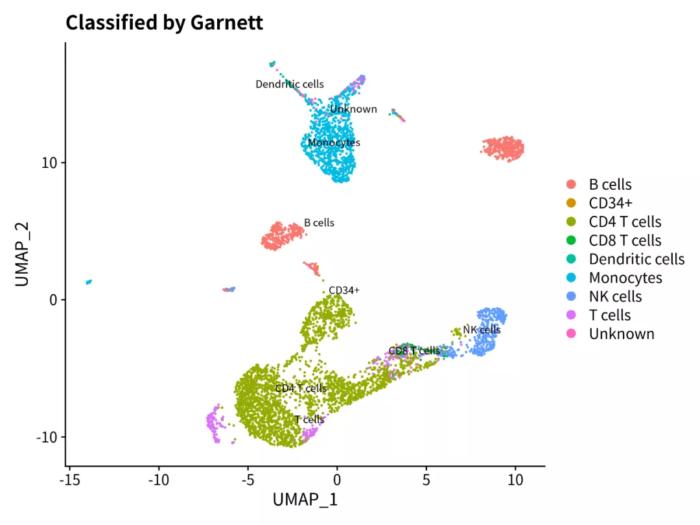
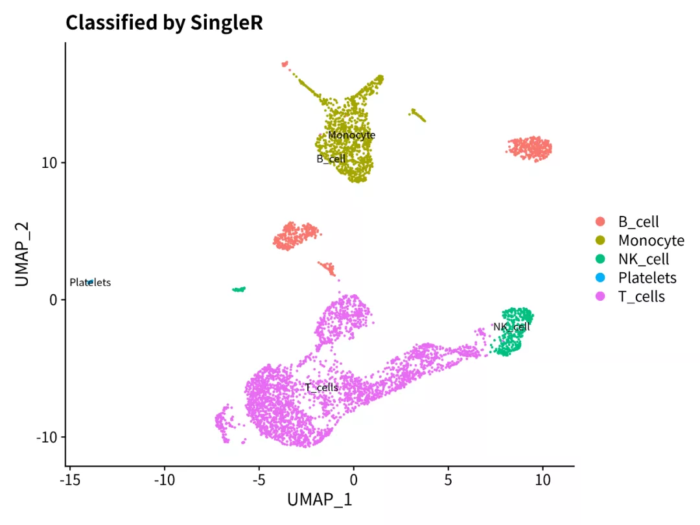
三种结果对比
- table(pbmc$cluster_ext_type)
- # B cells CD34 CD4 T cells CD8 T cells Dendritic cells Monocytes NK cells
- # 625 12 2484 81 73 945 486
- # T cells Unknown
- # 292 102
- table(pbmc$predicted.id)
- # ASDC B intermediate B memory B naive CD14 Mono CD16 Mono
- # 3 178 122 312 896 91
- # CD4 CTL CD4 Naive CD4 Proliferating CD4 TCM CD4 TEM CD8 Naive
- # 55 952 3 793 143 138
- # CD8 TCM CD8 TEM cDC1 cDC2 dnT Eryth
- # 29 262 4 61 7 373
- # gdT HSPC ILC MAIT NK NK Proliferating
- # 96 7 3 24 376 4
- # NK_CD56bright pDC Plasmablast Platelet Treg
- # 28 28 3 30 79
- table(pbmc$SingleR)
- # B_cell Monocyte NK_cell Platelets T_cells
- # 659 1085 386 30 2940
- p <- DimPlot(pbmc, group.by = "cluster_ext_type", label = T,
- label.size = 3) ggtitle("Classified by Garnett")
- ggsave("Garnett.png", p, width = 8, height = 6)
- p <- DimPlot(pbmc, group.by = "predicted.id", label = T,
- label.size = 3) ggtitle("Classified by Azimuth")
- ggsave("Azimuth.png", p, width = 8, height = 6)
- p <- DimPlot(pbmc, group.by = "SingleR", label = T,
- label.size = 3) ggtitle("Classified by SingleR")
- ggsave("SingleR.png", p, width = 8, height = 6)