背景知识
Doublets及其形成的原因
单细胞测序期望每个barcode标签下只有一个真实的细胞,但是实际数据中会有两个或多个细胞共用一个barcode的情况,业内称之为doublets或multiplets(后面统称为doublets)。Doublets形成的原因主要是高通量单细胞测序一般使用液滴微流控(droplet microfluidic)或纳米孔(nanowell)技术,细胞被液滴或纳米孔捕获的概率遵循泊松分布规律,doublets填充液滴的概率会随着输入细胞浓度升高而增加。此外,使用磁珠分选细胞,操作不当也会增加doublets形成的概率。
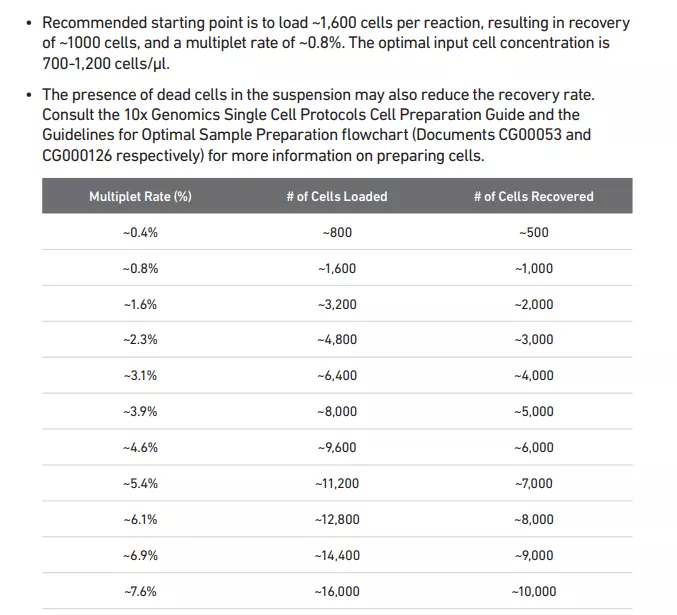
10X genomics单细胞平台的dulblets比率
CellRanger可以检测doublets吗?
10X genomics在官网对此问题进行过回复,大意如下:
CellRanger目前并不支持单物种样本doublets的检测,对于混合物种的样本(例如人和小鼠细胞1:1混合)可以根据基因组比对情况识别同时包含两个物种细胞的doublets(multiplets),也就是说cellranger检测doublets的能力聊胜于无。
10X genomics公司目前也没有官方推荐的dedoublets方法,但是官网的技术支持给出了三点建议:
看细胞内是不是有多种不应该在一起的marker基因,例如一个“细胞”或cluster中同时表达T细胞和B细胞的marker基因。 看“细胞”或cluster的基因数和UMI数是否异常,通常来说doublets会有更高的基因数和UMI数。 使用DoubletFinder之类的软件分析doublets。需要注意的是,dedoublets还没有权威方法,这些方法可能会过滤一些真正的单细胞。原文链接:https://kb.10xgenomics.com/hc/en-us/articles/360001074271-Does-Cell-Ranger-automatically-exclude-doublets-
综上所述:按目前8000 cells/样本的普遍上样量,doublets的问题确实值得重视。
DoubletFinder简介
分析原理
依据单细胞表达矩阵建立的低维空间中,表达特征相似的细胞彼此之间距离更近。DoubletFinder生成人工模拟的doublets,并将他们掺入原始单细胞表达数据,原则上人工模拟的doublets会与真实的doublets距离较近。因此计算每个细胞K最近邻细胞中人工模拟doublets的比例(pANN),就可以根据pANN值对每个barcode的doublets概率进行排序。另外依据泊松分布的统计原理可以计算每个样本中doublets的数量,结合之前的细胞pANN值排序,就可以过滤doublets了。需要注意的是DoubletFinder对相同细胞类型构成的doublets不敏感,因为这些细胞在表达特征上与真实的单细胞没有明显的差异。
分析流程
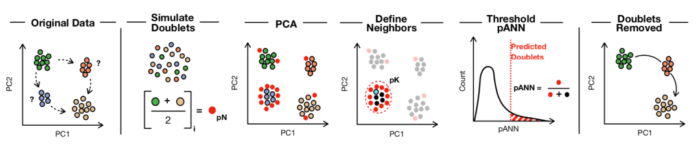
实际分析中主要有以下四步:
使用单细胞数据创建seurat对象,并进行数据标准化、降维,最好进行聚类和细胞类型鉴定; 使用BCmvn(均值-方差标准化双峰系数)寻找计算pANN的最优pK值; 根据泊松分布统计原理估计样本中doublets的数量,并排除DoubletFinder不能检出的同源doublets,得到优化后的预估doublets数量; 使用前两步得到的参数运行函数鉴定doublets数据。
软件安装
- remotes::install_github('chris-mcginnis-ucsf/DoubletFinder')
10X PBMC数据实测
测试数据来源于10x genomics官网的示例数据集。
数据下载链接:https://cf.10xgenomics.com/samples/cell-exp/3.0.2/5k_pbmc_v3_nextgem/5k_pbmc_v3_nextgem_filtered_feature_bc_matrix.h5
保存文件名:pbmc.h5
deDoulblets代码
以下代码使用SCT标准化方法,如果使用log标准化,把所有函数中的sct参数设置为 sct = F 即可。
- library(DoubletFinder)
- library(tidyverse)
- library(Seurat)
- library(patchwork)
- rm(list=ls())
- dir.create("DoulbletFinder")
- setwd("DoulbletFinder")
- ## 下载数据并预处理
- pbmc <- Read10X_h5("pbmc.h5")
- pbmc <- CreateSeuratObject(pbmc, min.cells = 3, min.features = 200)
- pbmc <- SCTransform(pbmc)
- pbmc <- RunPCA(pbmc, verbose = F)
- ElbowPlot(pbmc)
- pc.num=1:15
- pbmc <- RunUMAP(pbmc, dims=pc.num)
- pbmc <- FindNeighbors(pbmc, dims = pc.num) %>% FindClusters(resolution = 0.3)
- ## 寻找最优pK值
- sweep.res.list <- paramSweep_v3(pbmc, PCs = pc.num, sct = T)
- sweep.stats <- summarizeSweep(sweep.res.list, GT = FALSE)
- bcmvn <- find.pK(sweep.stats)
- pK_bcmvn <- bcmvn$pK[which.max(bcmvn$BCmetric)] %>% as.character() %>% as.numeric()
- ## 排除不能检出的同源doublets,优化期望的doublets数量
- DoubletRate = 0.039 # 5000细胞对应的doublets rate是3.9%
- homotypic.prop <- modelHomotypic(pbmc$seurat_clusters) # 最好提供celltype
- nExp_poi <- round(DoubletRate*ncol(pbmc))
- nExp_poi.adj <- round(nExp_poi*(1-homotypic.prop))
- ## 使用确定好的参数鉴定doublets
- pbmc <- doubletFinder_v3(pbmc, PCs = pc.num, pN = 0.25, pK = pK_bcmvn,
- nExp = nExp_poi.adj, reuse.pANN = F, sct = T)
- ## 结果展示,分类结果在pbmc@meta.data中
- DimPlot(pbmc2, reduction = "umap", group.by = "DF.classifications_0.25_0.3_171")
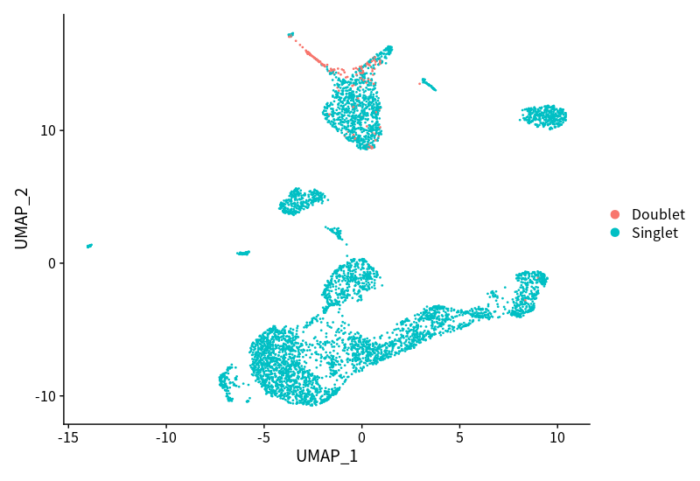
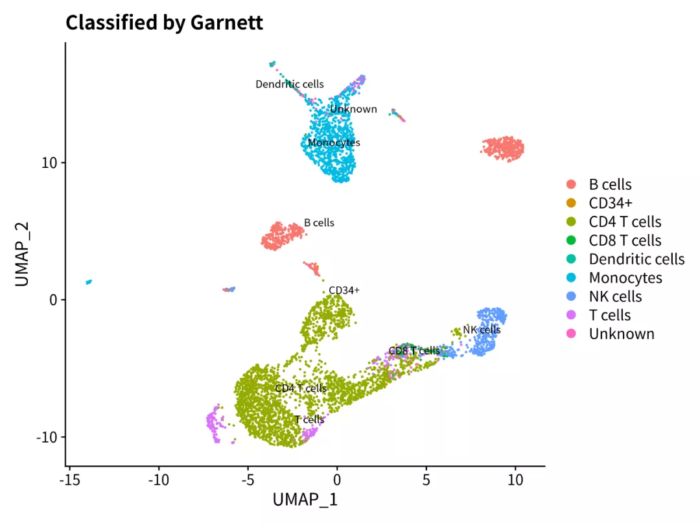
对比《单细胞分析之Garnett》中使用Gernett分类器鉴定的结果(下图),我发现一个有意思的现象:DoubletFinder识别的doublets与Garnett定义为Unknown的类型有一些是重合的。