

NMF算法简介
NMF是什么?
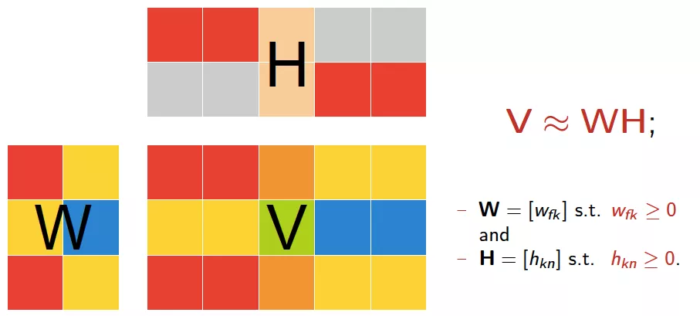
非负矩阵分解(Non-negative Matrix Factorization, NMF)本质上说是一种矩阵分解的方法,对于任意给定的一个非负矩阵V,NMF算法能够寻找到一个非负矩阵W和一个非负矩阵H,使得 V≈W*H成立 ,从而将一个非负的矩阵分解为左右两个非负矩阵的乘积。
NMF的特点?
NMF最重要的特点是非负性约束。矩阵分解的方法有很多种,如奇异值分解 (singular value decomposition, SVD) 、独立成分分析 (independent component analysis, ICA) 、主成分分析 (principal component analysis, PCA) 等。这些方法的共同特点是,即使初始矩阵 V 元素是非负的,分解出来的因子 W 和 H 中的元素往往含有负值元素。从计算科学的角度来看,分解出来的因子 W 和 H 中的元素含有负值元素并没有问题, 但负值元素通常是无法解释的。NMF约束了原始矩阵V和分解矩阵W、H的非负性,这就意味着只能通过特征的相加来实现原始矩阵V的还原,最终导致的结果是:
非负性会引发稀疏 非负性会使计算过程进入部分分解
给大家对比一下PCA与NMF分解图像的效果:
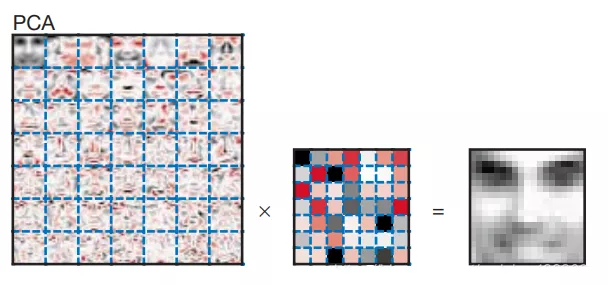
PCA分解之后,每个主成分(PC)都会保留与其他PC不正交的全局特征,并且PC保留的特征是递减的。
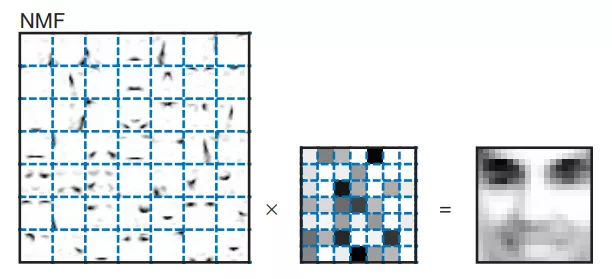
NMF分解之后,每个因子保留的都是局部特征,它们的权重是基本平等的。通过这张图可以看出,很多因子能与面部特征一一对应起来,例如鼻子、眼睛、嘴巴都能找到相应的因子。
NMF在单细胞研究中的优势
单细胞研究避免不了要回答两个问题:组织中有哪些细胞类型,每个细胞类型又有哪些表达模式?NMF解决这类问题具有天然的优势,因为它分解的因子很容易与细胞类型或表达模式对应起来。Github上有很多基于NMF和其变种算法的单细胞分析工具,我比较喜欢的有单细胞整合分析工具liger和空间转录组去卷积工具SPOTlight。应用NMF分析方法发表的高分文章也有很多,我给大家介绍一篇,更多的文章请自己搜索。
Chen, YP., Yin, JH., Li, WF. et al. Single-cell transcriptomics reveals regulators underlying immune cell diversity and immune subtypes associated with prognosis in nasopharyngeal carcinoma. Cell Res 30, 1024–1042 (2020). https://doi.org/10.1038/s41422-020-0374-x
基础NMF包的安装与用法简介
安装NMF基础包
- BiocManager::install('Biobase')
- install.packages('NMF')
nmf函数简介
NMF包通过nmf()函数实现矩阵分解,它的用法及重要参数如下:
nmf(x, rank, method, seed, nrun, ...)
x:待分解非负矩阵,数据格式可以是matrix,data.frame, ExpressionSet
rank:分解的基数量,对于单细胞数据,可以设置为期望的细胞类型数量或表达模式数量
method:因式分解的常用方法,这里介绍三种常用的
1、基于KL 散度进行度量目标函数的多重迭代梯度下降算法——brunet(默认算法)
2、基于欧几里得距离度量目标函数的多重迭代梯度下降算法——lee
3、交替最小二乘法(Alternating Least Squares(ALS))——snmf/r
seed:因式分解的初始化种子
nrun:运行次数
rank值一般是要通过测试评估后确定的,但是分析单细胞数据这是一个很难完成的工作,5000个细胞的测试时间可能超过10个小时。替代办法是使用经验或先验知识指定,可以尝试略多于细胞类型或细胞状态(细胞亚群再聚类时)的一个数值,例如我在本帖的PBMC数据分解中就指定为rank=10。因为NMF一般是从随机数开始,通过迭代算法收敛误差的方法求出最优W和H矩阵,所以seed不同最后的结果也不同。为了减少seed的影响求得最优解,常规的办法是通过nrun参数设置运行100-200次矩阵分解选取最优值,也可以使用特殊的算法选择一个最佳的seed(设置seed='nndsvd'或seed='ica'),这样运行一次也能得到最优解。下面我们测试一下不同方法的运行时间:
- library(Biobase)
- library(NMF)
- ## 参数测试
- data("esGolub")
- #esGolub <- esGolub[1:500,]
- t1 <- nmf(esGolub, 3, method = "brunet", seed = 219)
- runtime(t1) # elapsed: 1.239
- t2 <- nmf(esGolub, 3, method = "lee", seed = 219)
- runtime(t2) # elapsed: 1.471
- t3 <- nmf(esGolub, 3, method = "snmf/r", seed = 219)
- runtime(t3) # elapsed: 0.995
- t4 <- nmf(esGolub, 3, method = "brunet", seed = 'nndsvd')
- runtime(t4) # elapsed: 3.156
- t5 <- nmf(esGolub, 3, method = "brunet", nrun = 100)
- runtime(t5) # elapsed: 2.363
10X PBMC数据实测
测试数据
测试数据来源于10x genomics官网的示例数据集。
数据下载链接:https://cf.10xgenomics.com/samples/cell-exp/3.0.2/5k_pbmc_v3_nextgem/5k_pbmc_v3_nextgem_filtered_feature_bc_matrix.h5
保存文件名:pbmc.h5
基于PCA分解的降维聚类
- library(Seurat)
- library(tidyverse)
- library(NMF)
- rm(list = ls())
- ## 创建seurat对象
- pbmc <- Read10X_h5("pbmc.h5")
- pbmc <- CreateSeuratObject(pbmc, project = "pbmc", min.cells = 3, min.features = 500)
- pbmc$percent.mt <- PercentageFeatureSet(pbmc, pattern = "^MT-")
- pbmc <- subset(pbmc, percent.mt<20)
- pbmc <- NormalizeData(pbmc) %>% FindVariableFeatures() %>% ScaleData(do.center = F)
- ## 使用pca的分解结果降维聚类
- pbmc <- RunPCA(pbmc)
- set.seed(219)
- pbmc.pca <- RunUMAP(pbmc, dims = 1:15) %>% FindNeighbors(dims = 1:15) %>% FindClusters()
- ## 结果可视化
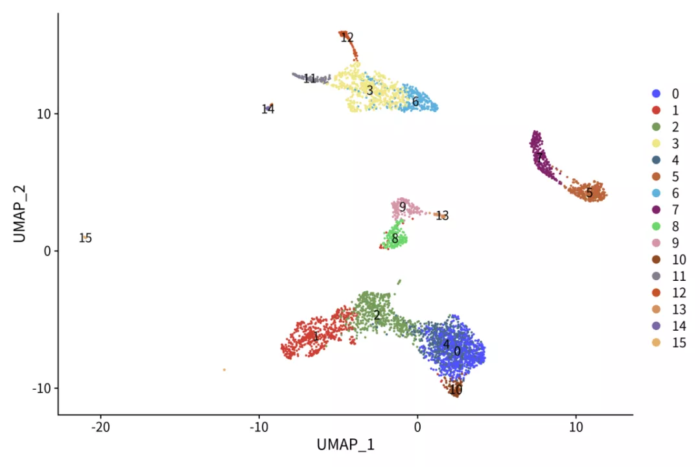
- p <- DimPlot(pbmc.pca, label = T) ggsci::scale_color_igv()
- ggsave("pbmc_pca.png", p, width = 9, height = 6)
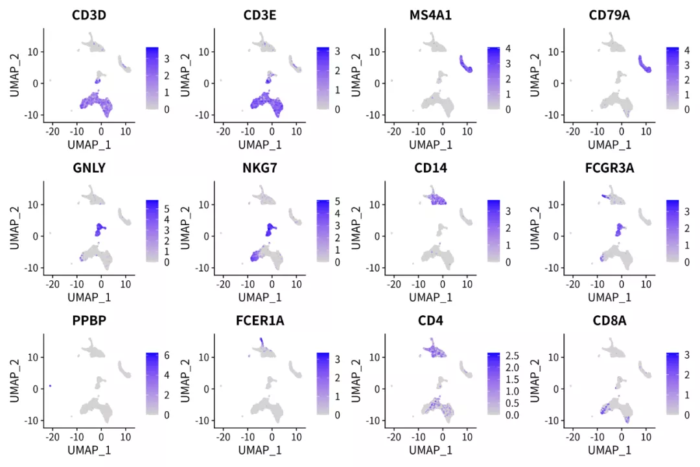
- p <- FeaturePlot(pbmc.pca, features = c('CD3D', 'CD3E', 'MS4A1', 'CD79A', 'GNLY', 'NKG7', 'CD14',
- 'FCGR3A', 'PPBP', 'FCER1A', 'CD4', 'CD8A'), ncol = 4)
- ggsave("pbmc_pca_markers.png", p, width = 12, height = 8)
基于NMF分解的降维聚类
- ## 高变基因表达矩阵的分解
- # pbmc大体可分成T,B,NK,CD14 Mono,CD16 Mono,DC,Platelet等类型,考虑冗余后设置rank=10
- vm <- pbmc@assays$RNA@scale.data
- res <- nmf(vm, 10, method = "snmf/r", seed = 'nndsvd')
- runtime(res)
- # 用户 系统 流逝
- #1063.147 78.019 1139.831
- ## 分解结果返回suerat对象
- pbmc@reductions$nmf <- pbmc@reductions$pca
- pbmc@reductions$nmf@cell.embeddings <- t(coef(res))
- pbmc@reductions$nmf@feature.loadings <- basis(res)
- ## 使用nmf的分解结果降维聚类
- set.seed(219)
- pbmc.nmf <- RunUMAP(pbmc, reduction = 'nmf', dims = 1:10) %>%
- FindNeighbors(reduction = 'nmf', dims = 1:10) %>% FindClusters()
- ## 结果可视化
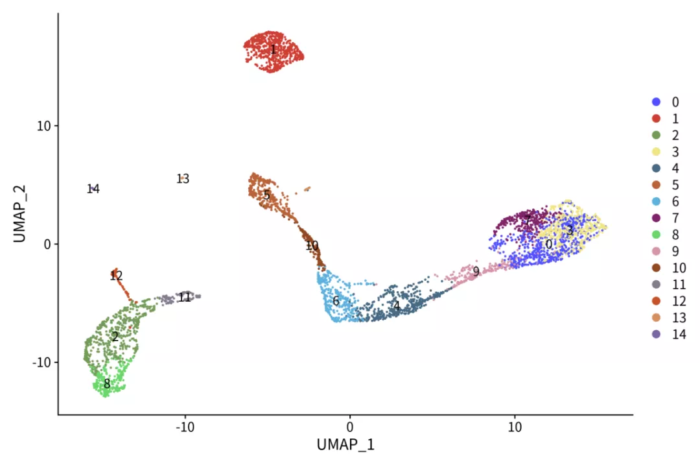
- p <- DimPlot(pbmc.nmf, label = T) ggsci::scale_color_igv()
- ggsave("pbmc_nmf.png", p, width = 9, height = 6)
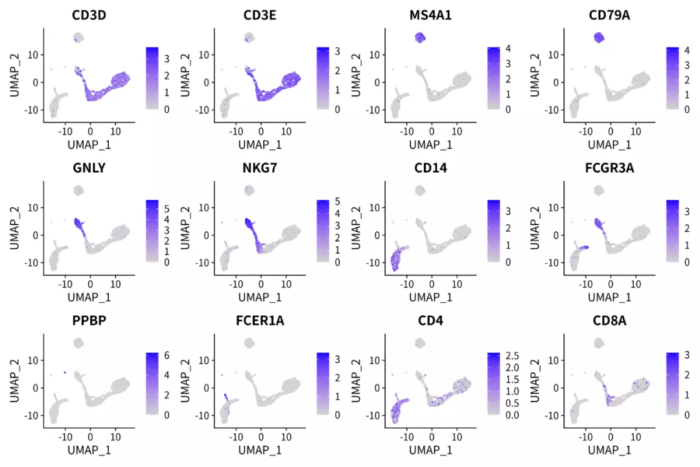
- p <- FeaturePlot(pbmc.nmf, features = c('CD3D', 'CD3E', 'MS4A1', 'CD79A', 'GNLY', 'NKG7', 'CD14',
- 'FCGR3A', 'PPBP', 'FCER1A', 'CD4', 'CD8A'), ncol = 4)
- ggsave("pbmc_nmf_markers.png", p, width = 12, height = 8)
NMF因子可解释性探索
对比PCA分析的结果,NMF虽然毫不逊色,但是它的运行时间更长,我们为什么要用NMF呢?一个很重要的原因是NMF的因子可解释性更强,每个因子贡献度最大的基因基本代表了某种或某个状态细胞的表达模式,相比差异分析得到marker基因更有代表性。
NMF因子与细胞类型的关系
- ## 人工定义细胞类型
- pbmc.nmf$celltype <- pbmc.nmf$seurat_clusters
- pbmc.nmf$celltype <- recode(pbmc.nmf$celltype,
- '1' = "B cells",
- '5' = "NKs",
- '10' = "CD8 T",
- '6' = "CD8 T",
- '4' = "CD4 T",
- '9' = "CD4 T",
- '0' = "CD4 T",
- '3' = "CD4 T",
- '7' = "CD4 T",
- '2' = "CD14 Mono",
- '8' = "CD14 Mono",
- '11' = "CD16 Mono",
- '12' = "DCs",
- '13' = "Platelet",
- '14' = "Unknown")
- p <- DimPlot(pbmc.nmf, group.by = 'celltype', label = T, label.size = 3) ggsci::scale_color_npg(alpha = 0.6)
- ggsave("pbmc_nmf_celltype.png", p, width = 9, height = 6)
- ## 查看细胞因子上的荷载
- tmp <- data.frame(t(coef(res)), check.names = F)
- colnames(tmp) <- paste0("factor", 1:10)
- pbmc.nmf <- AddMetaData(pbmc.nmf, metadata = tmp)
- p <- FeaturePlot(pbmc.nmf, features = paste0("factor", 1:10), ncol = 4)
- ggsave("pbmc_nmf_factors.png", p, width = 12, height = 8)
- ## 查看细胞主成分上的荷载
- p <- FeaturePlot(pbmc.nmf, features = paste0("PC_", 1:12), ncol = 4)
- ggsave("pbmc_nmf_PCs.png", p, width = 12, height = 8)
人工鉴定的细胞类型
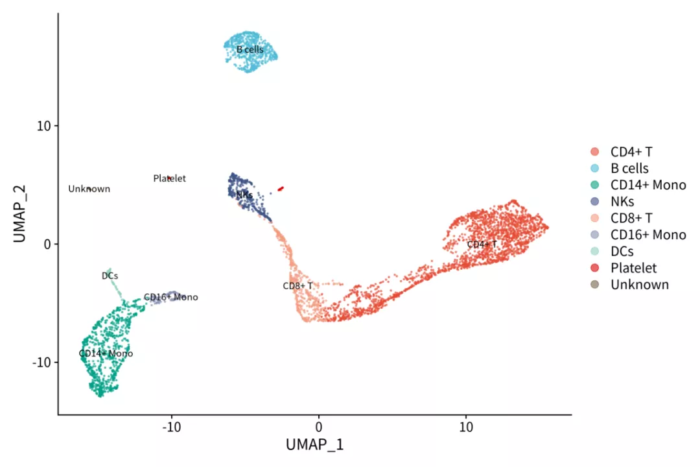
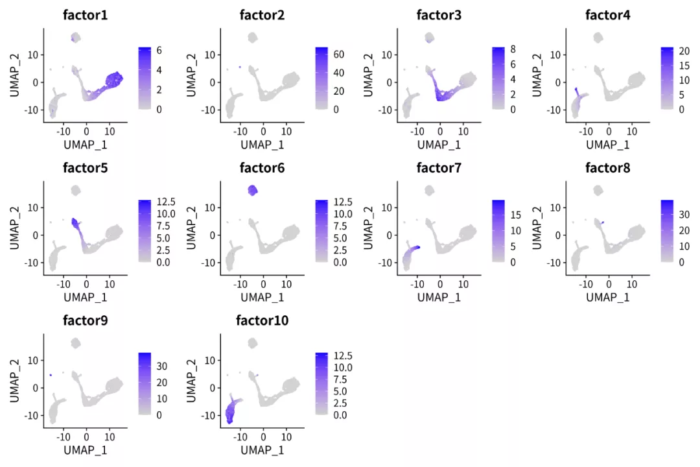
细胞在因子上的值
 细胞在PC轴上的值
细胞在PC轴上的值
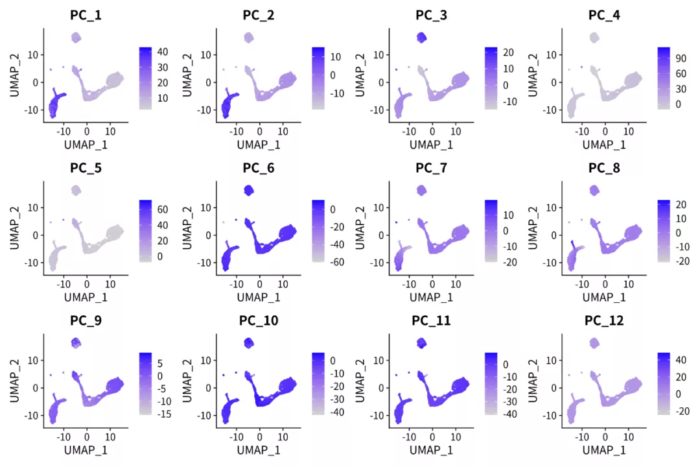
对比上下两张图,很容易发现NMF的因子比PCA的PC轴解释性更强。
提取celltype的signatures
- ## 提取每个因子贡献度最大的20个基因
- f <- extractFeatures(res, 20L)
- f <- lapply(f, function(x) rownames(res)[x])
- f <- do.call("rbind", f)
- DT::datatable(t(f))
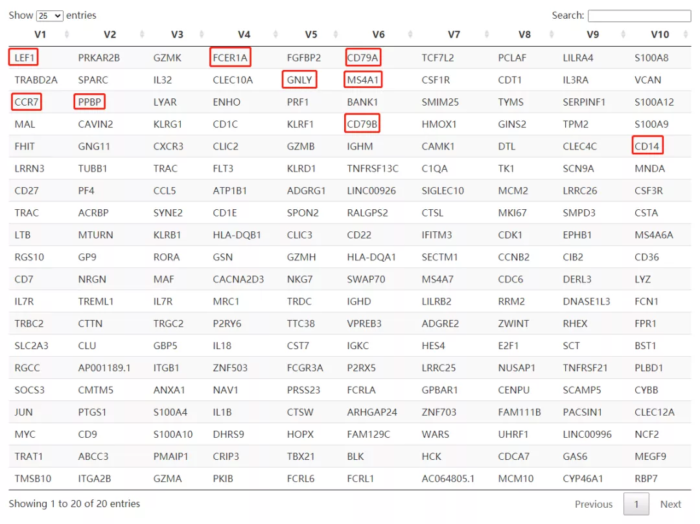
NMF的10个因子中,很容易发现我们对细胞进行分类的marker基因,是不是很神奇?