

xCell简介
xCell是开发SingleR包的团队2017年推出的一款推断bulkRNA样本中细胞类型比例的R包,目前在google学术查到它有598次引用。xCell的工作原理是用机器学习算法提取了64种免疫细胞和基质细胞的signature,待检测bulkRNA数据先用ssGSEA算法计算样本在每个细胞类型signature的富集分数,然后用特别设计的算法将样本中各种细胞类型的富集分数转换为细胞类型分数,最后对紧密相关的细胞类型分数进行补偿校正。
xCell支持的64种细胞类型
xCell使用须知
使用xCell分析的bulkRNA数据类型可以是RNA-seq测序数据,也可以是表达芯片数据,但不要把两种数据混合在一起分析。表达芯片数据不用对数据进行任何处理,测序数据要转换为TPM、FPKM或RPKM值。 如果输入样本中的细胞成分没有足够的可变性,xCell将无法识别任何信号;因此输入数据必须具有异质性,且不要把多个样本分成多次运行xCell,不同运行之间的输出结果没有可比性。 xCell中的线性变换使用校准参数使xCell评分与百分比相似,但是它有时也会不准确,如果分析得出的高分对应明显错误的细胞类型,可以手动调整校准参数。 xCell评分是基于signatures的富集分数,它与真正的细胞比例有线性相关性,但是不能把xCell评分作为细胞比例值。xCell评分用于下游分析时,可以在不同样本之间对比同一细胞类型的得分,但是不要在同一样本内比较不同细胞类型的得分。 不要把xCell用于单细胞数据的细胞类型鉴定。
xCell用法
示例数据
- https://raw.githubusercontent.com/dviraran/xCell/master/vignettes/sdy420.rds
安装xCell
- devtools::install_github('dviraran/xCell')
xCell测试
- library(xCell)
- ## 加载测试数据
- sdy <- readRDS("sdy420.rds")
- # sdy是下载的示例数据,有104个样本的表达谱芯片的bulkRNA数据expr,
- # 以及基于流式计数的细胞百分比的数据fcs
- summary(sdy)
- # Length Class Mode
- #expr 104 data.frame list
- #fcs 104 data.frame list
- ## 根据样本实际情况设置分析的细胞类型,有利于提高分析的准确性,非必要步骤
- cell.types.use = intersect(colnames(xCell.data$spill$K), rownames(sdy$fcs))
- ## xCell评分,注意rnaseq参数,芯片数据设为F,测序数据设为T
- scores = xCellAnalysis(sdy$expr, rnaseq=F, cell.types.use = cell.types.use)
- ## 准确性评估
- library(psych)
- library(ggplot2)
- fcs = sdy$fcs[rownames(scores), colnames(scores)]
- res = corr.test(t(scores), t(fcs), adjust='none')
- qplot(x=rownames(res$r), y=diag(res$r),
- fill=diag(res$p) < 0.05, geom='col',
- main='SDY420 association with immunoprofiling',
- ylab='Pearson R', xlab= '') labs(fill = "p-value < 0.05")
- theme_classic()
- theme(axis.text.x = element_text(angle = 45, hjust = 1))
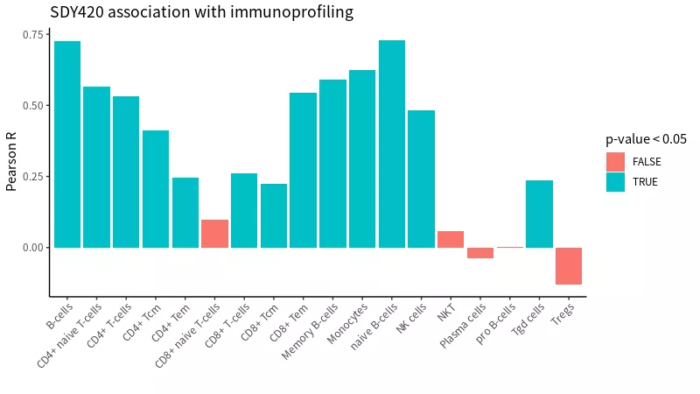
测试数据集中有24种细胞类型,因为xCell自带signature的局限性,只能预测18种细胞类型。其中13个预测结果与真实的细胞比例显著相关(p<0.05),7个预测结果与真实的细胞比例高度相关(p<0.05&R>0.5)。
关于介绍 xCell 的说明
xCell并不是一款分析单细胞数据的工具,我向大家介绍它并收录在《单细胞分析十八般武艺》专题中,是因为它与单细胞的分析密切相关。虽然单细胞研究热潮已经持续了几年,但是高昂的成本依然让大家难以负担;因此使用少量样本做scRNA-seq得出研究结论,然后用大量bulkRNA样本进行验证的策略被越来越多的人使用。为了更好地将单细胞数据与bulkRNA数据联系起来,往往需要对bulkRNA数据进行去卷积操作,近年来很多优秀的去卷积工具被开发出来,我会在此专题中陆续介绍几款常用的方法。