

Control-FREEC是一款广受好评的WGS、WES数据CNV检测软件。
安装
当前最新版本是v11.6(29 May 2020)。
下载源码编译安装
- wget https://github.com/BoevaLab/FREEC/archive/refs/tags/v11.6.tar.gz
- tar -zxvf v11.6.tar.gz
- cd FREEC-11.6/src
- make
获得编译后的freec执行程序。
如果需要统计GC比例,还需要安装gccount
- wget http://bioinfo-out.curie.fr/projects/freec/src/gccount.tar.gz
- tar -zxvf gccount.tar.gz -C gccount
gccount已预编译好,解压后可直接使用。
另外,control-freec需要环境中包含(或配置文件中指定路径)R、samtools、bedtools、sambamba等。其中samtools和sambamba用于处理bam文件,bedtools用于生成minipileup格式文件。
如果需要结果中包含比对信息,需下载mappability track,官方提供hg19、hg38、mm9、mm10等基因组的track。
如果需要检测等位染色体状态,需下载dbsnp文件,官方提供了hg19、hg18、mm10等,不知道和ncbi提供的dbsnp格式是否存在差异。
使用
Control-Freec的使用命令非常简单
- freec -conf conf.txt
重要的是配置文件,软件源码库中包含了WES和WGS等两个例子。但是,例子是的内容是相当不全的,更多的配置标签需要看官网的教程。
以下是WGS例子,我增加了输出路径等及删除了部分用不上的参数。
- [general]
- outputDir = /path/to/output
- chrLenFile = b37.freec.len
- ploidy = 2
- breakPointThreshold = .8
- #coefficientOfVariation = 0.01
- window = 50000
- #step=10000
- chrFiles = /path/to/b37/sep_chrom
- GCcontentProfile = /path/to/b37/b37.freec.50kb.gc.cnp
- maxThreads = 8
- #readCountThreshold=10
- #numberOfProcesses = 4
- #outputDir = test
- #contaminationAdjustment = TRUE
- #contamination = 0.4
- #minMappabilityPerWindow = 0.95
- #breakPointType = 4
- #forceGCcontentNormalization = 0
- sex=XY
- #BedGraphOutput=TRUE
- sambamba = sambamba
- SambambaThreads = 8
- [sample]
- mateFile = test.bam
- #mateCopyNumberFile = test/sample.cpn
- inputFormat = BAM
- mateOrientation = 0
- ##use "mateOrientation=0" for sorted .SAM and .BAM
- [control]
- #mateFile = /path/control.pileup.gz
- #mateCopyNumberFile = path/control.cpn
- #inputFormat = pileup
- #mateOrientation = RF
- #[BAF]
- ##use the following options to calculate B allele frequency profiles and genotype status. This option can only be used if "inputFormat=pileup"
- #SNPfile = /bioinfo/users/vboeva/Desktop/annotations/hg19_snp131.SingleDiNucl.1based.txt
- #minimalCoveragePerPosition = 5
- ##use "minimalQualityPerPosition" and "shiftInQuality" to consider only high quality position in calculation of allelic frequencies (this option significantly slows down reading of .pileup)
- #minimalQualityPerPosition = 5
- #shiftInQuality = 33
- [target]
- ##use a tab-delimited .BED file to specify capture regions (control dataset is needed to use this option):
- #captureRegions = /bioinfo/users/vboeva/Desktop/testChr19/capture.bed
后面的target标签应该是做靶向捕获数据或者只想分析某个区域数据时才需要的,而BAF标签则是需要分析LOH等内容时才需要。要进行BAF分析时,输入文件格式必须时pileup而不是bam。
general标签中的chrLenFile需要的是染色体的长度,可以直接使用samtools faidx 生成的fai索引(未测试),官网也提供了hg19的这个文件。如果是使用GATK的b37参考基因组,则把chr去掉即可。
chrFiles需要填的是fasta格式文件的路径,建议是该路径下仅包含reference的各个染色体的fasta文件,不要包含其他基因组的fasta文件,命名要与chrLenFile中的染色体名相同。如chr1.fa、chr2.fa等。
可以使用samtools对参考基因组进行提取,把每个染色体拆分出来
- samtools faidx hg19.fa chr1 > seq_chrom/chr1.fa
GCcontentProfile则是需要划分的每个bin的GC比例用于校正,获得的方法是使用gccount软件,conf.txt文件与freec的用同一个即可。
- gccount -conf conf.txt
后续分析
软件的速度还是比较快的,上述分析完成后,会生成后缀为_CNVs、_ratio.txt、_sample.cpn、_info.txt等文件。结果在CNVs文件中。
再使用assess_significance.R脚本补充p值,这个脚本需要安装R包“rtracklayer”
- cat FREEC-11.6/scripts/assess_significance.R | R --slave --args test.bam_CNVs test.bam_ratio.txt
可使用makeGraph.R画图
- cat FREEC-11.6/scripts/makeGraph.R | R --slave --args 2 test.bam_ratio.txt
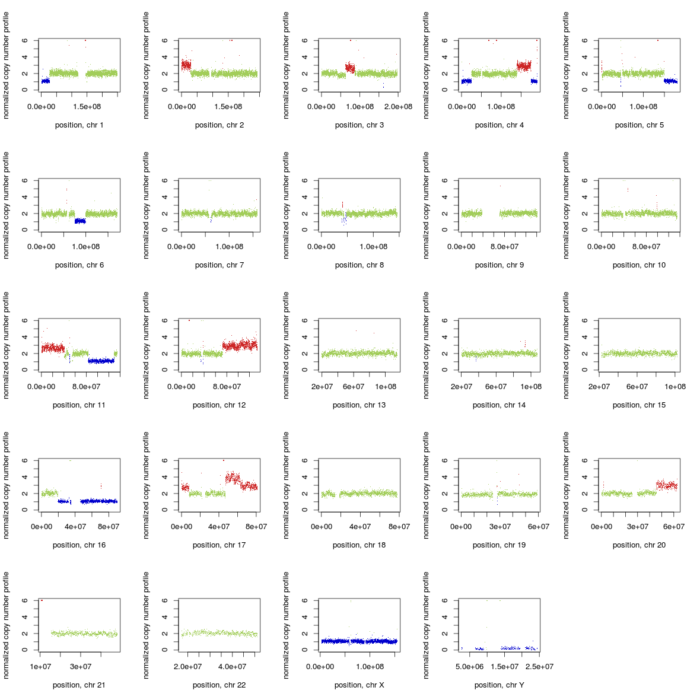
将ratio文件转为bed格式或circos格式
- perl FREEC-11.6/scripts/freec2bed.pl -f test.bam_ratio.txt > test.bam_ratio.bed
- perl FREEC-11.6/scripts/freec2circos.pl -f test.bam_ratio.txt > test.bam_ratio.circos