

显著性检验通常可以告诉我们一个观测值是否是有效的,例如检测两组样本均值差异的假设检验可以告诉我们这两组样本的均值是否相等(或者那个均值更大)。我们在实验中经常会因为各种问题(时间、经费、人力、物力)得到一些小样本结果,如果我们想知道这些小样本结果的总体是什么样子的,就需要用到置换检验。
Permutation test 置换检验是Fisher于20世纪30年代提出的一种基于大量计算(computationally intensive),利用样本数据的全(或随机)排列,进行统计推断的方法,因其对总体分布自由,应用较为广泛,特别适用于总体分布未知的小样本资料,以及某些难以用常规方法分析资料的假设检验问题。在具体使用上它和Bootstrap Methods类似,通过对样本进行顺序上的置换,重新计算统计检验量,构造经验分布,然后在此基础上求出P-value进行推断。
下面通过一个简单例子来介绍Permutation test的思想。
假设我们设计了一个实验来验证加入某种生长素后拟南芥的侧根数量会明显增加。A组是加入某种生长素后,拟南芥的侧根数量;B是不加生长素时,拟南芥的侧根数量(均为假定值)。
A组侧根数量(共12个数据):24 43 58 67 61 44 67 49 59 52 62 50
B组侧根数量(共16个数据):42 43 65 26 33 41 19 54 42 20 17 60 37 42 55 28
我们来用假设检验的方法来判断生长素是否起作用。我们的零假设为:加入的生长素不会促进拟南芥的根系发育。在这个检验中,若零假设成立,那么A组数据的分布和B组数据的分布是一样的,也就是服从同个分布。
接下来构造检验统计量——A组侧根数目的均值同B组侧根数目的均值之差。
statistic:= mean(Xa)-mean(Xb)
对于观测值有 Sobs:=mean(Xa)-mean(Xb)=(24+43+58+67+61+44+67+49+59+52+62+50)/12-(42+43+65+26+33+41+19+54+42+20+17+60+37+42+55+28)/16=14
我们可以通过Sobs在置换分布(permutation distribution)中的位置来得到它的P-value。
Permutation test的具体步骤是:
1.将A、B两组数据合并到一个集合中,从中挑选出12个作为A组的数据(X'a),剩下的作为B组的数据(X'b)。
Gourp:=24 43 58 67 61 44 67 49 59 52 62 50 42 43 65 26 33 41 19 54 42 20 17 60 37 42 55 28
挑选出 X'a:=43 17 44 62 60 26 28 61 50 43 33 19
X'b:=55 41 42 65 59 24 54 52 42 49 37 67 67 20 42 58
2.计算并记录第一步中A组同B组的均值之差。Sper:=mean(X'a)-mean(X'b)= -7.875
3.对前两步重复999次(重复次数越多,得到的背景分布越”稳定“)
这样我们得到有999个置换排列求得的999个Sper结果,这999个Sper结果能代表拟南芥小样本实验的抽样总体情况。
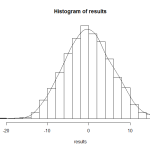
permutation test
如上图所示,我们的观测值 Sobs=14 在抽样总体右尾附近,说明在零假设条件下这个数值是很少出现的。在permutation得到的抽样总体中大于14的数值有9个,所以估计的P-value是9/999=0.01
最后还可以进一步精确P-value结果(做一个抽样总体校正),在抽样总体中加入一个远大于观测值 Sobs=14的样本,最终的P-value=(9+1)/(999+1)=0.01。(为什么这样做是一个校正呢?自己思考:))
结果表明我们的原假设不成立,加入生长素起到了促使拟南芥的根系发育的作用。
参考资料:
1. http://bcs.whfreeman.com/ips5e/content/cat_080/pdf/moore14.pdf
2. http://jpkc.njmu.edu.cn/course/tongjixue/file/jxzy/tjjz02.htm
3. http://www.r-bloggers.com/lang/chinese/541
附录:R语言求解上例的代码
- a<-c(24,43,58,67,61,44,67,49,59,52,62,50,42,43,65,26,33,41,19,54,42,20,17,60,37,42,55,28)
- group<-factor(c(rep("A",12),rep("B",16)))
- data<-data.frame(group,a)
- find.mean<-function(x){
- mean(x[group=="A",2])-mean(x[group=="B",2])
- }
- results<-replicate(999,find.mean(data.frame(group,sample(data[,2]))))
- p.value<-length(results[results>mean(data[group=="A",2])-mean(data[group=="B",2])])/1000
- hist(results,breaks=20,prob=TRUE)
- lines(density(results))
1F
在电脑上测试了下,果然不错,学习了
B1
@ xiayan 最后那里,为什么这样做是一个校正呢?
2F
非常好谢谢
3F
谢谢楼主,以前一直不明白permutation test是怎么做的,这下明白了。讲得很好。
4F
我有3个问题,
1.在第三步进行重复步骤时,是不是按照排列的计算方法来重复
如果是按照排列来计算的话,是不是本教程的第三应该改为30421755次
2.计算最后精确P-value时,是采用在在抽样总体中加入一个远大于观测值 Sobs=14的样本的方法来矫正的
想不明白
3.排列组合的定义为,从不含重复元素的集合中抽取元素来进行计算,那我们在实际应用时,是不是在把treat和control合并后的数据集进行去重?
问题有点多,感谢楼主的回答
B1
@ 沙卡 我看不懂你提出的第一个问题,第二个问题请自己思考什么是“样本”什么是“总体”,第三个问题也看不懂。我觉得你首先要明白这是一个“假设检验”问题,不是一个枚举问题。方法中用到了样本数据的置换,构造不同的观测结果,但不需要枚举全排列所有组合情况。
来自外部的引用