

一、 DAVID简介
DAVID (the Database for Annotation,Visualization and Integrated Discovery)的网址是http://david.abcc.ncifcrf.gov/。 DAVID是一个生物信息数据库,整合了生物学数据和分析工具,为大规模的基因或蛋白列表(成百上千个基因ID或者蛋白ID列表)提供系统综合的生物功能注释信息,帮助用户从中提取生物学信息。
DAVID这个工具在2003年发布,目前版本是v6.7。和其他类似的分析工具,如GoMiner,GOstat等一样,都是将输入列表中的基因关联到生物学注释上,进而从统计的层面,在数千个关联的注释中,找出最显著富集的生物学注释。最主要是功能注释和信息链接。
二、 分析工具
DAVID需要用户提供感兴趣的基因列表,在基因背景下,使用提供的分析工具,提取该列表中含有的生物信息。这里说的基因列表和背景文件的选取对结果至关重要。
1、基因列表:
这个基因列表可能是上游的生物信息分析产生的基因ID列表。对于富集分析而言,一般情况下,大量的基因组成的列表有更高的统计意义,对富集程度高的特殊Terms有更高的敏感度。富集分析产生的p-value在相同或者数量相同的基因列表中具有可比性。
DAVID对于基因列表的格式要求为每行一个基因ID或者是基因ID用逗号分隔开。基因列表的质量会直接影响到分析结果。这里定性给出好的基因列表应该具有的特点,一个好的基因列表至少要满足以下的大部分的要求:
(1) 包含与研究目的相关的大部分重要的基因(如标识基因)。
(2) 基因的数量不能太多或者太少,一般是100至10000这个数量级。
(3) 大部分基因可以较好的通过统计筛选,例如,在控制组和对照组样品间选择显著差异表达基因时,使用的t-test标准:fold changes >=2 && P-values <=0.05。
(4) 大部分是上下调的基因都涉及到特定的某一生物过程,而不是随机的散布到所有可能的生物过程中。
(5) 一个好的基因列表比起随机产生的一个基因列表,应该含有更丰富的生物信息。
(6) 在同样的条件下,列表具有高度可重复性。
(7) 高通量数据的质量能够被其他独立的实验证实。
以上(2),(3),(6)&(7)是来自上游的数据标准,DAVID会自动检查其余的各项要求,即(1),(4)&(7)。
2、基因背景:
在一项研究中,如果一个生物过程不正常,那么通过高通量筛选技术,对该过程共同作用的基因有更大的可能性被选为相关的一组。富集分析正是以此为基础。为检测富集的程度,必须选取一个背景来进行对比。基因背景的选取有一个指导原则,就是必须构建一个足够大的,研究者可能涉及的所有基因的集合。用户使用默认的背景文件(默认为该物种的所有基因),或者是上传一个基因列表文件作为基因背景。
3、DAVID为实现各项功能分析,提供了以下4个分析内容(共6个分析工具):
(1) Gene Name Batch Viewer
这个工具能够实现将基因ID迅速翻译成基因名称,从而给研究者对于基因ID列表一个直观的印象,初步判断基因列表是否符合要求目的。
图1中显示了该工具的分析结果,具体说明图1中标注。
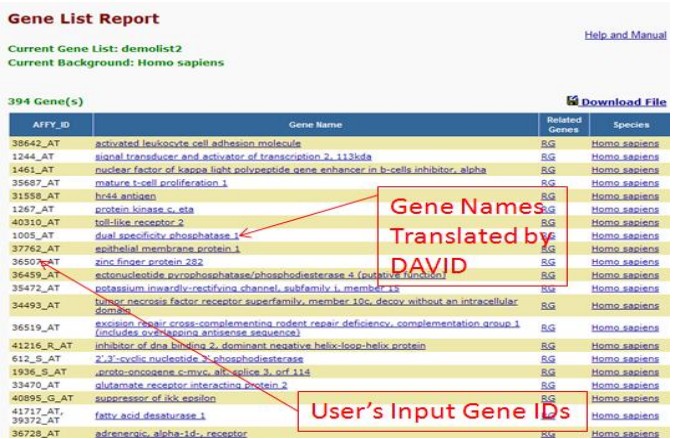
图1 Gene Name Batch Viewer的分析结果
(2) Gene Functional Classification
这个工具是Gene Name Batch Viewer工具的延伸。由于基因名称并不能显著体现基因的功能,所以我们需要更加有效的功能分类工具。该工具基于它们共同的注释信息,而不是基因名称,采用全新的模糊聚类算法,能够实现将功能相关的基因聚到一起作为一个单元,在生物学网络水平上去研究这些基因群。对聚类结果打分,分值越高,代表该组内的基因在基因列表中越重要。同时还提供了2-D View,以热图形式展现聚类到同一组的基因和该组内各个Term之间的关系。
结果见图2,将列表中的基因ID作为聚类对象,将功能相关的基因分组显示。图3是以热图形式展示的gene-term关系。
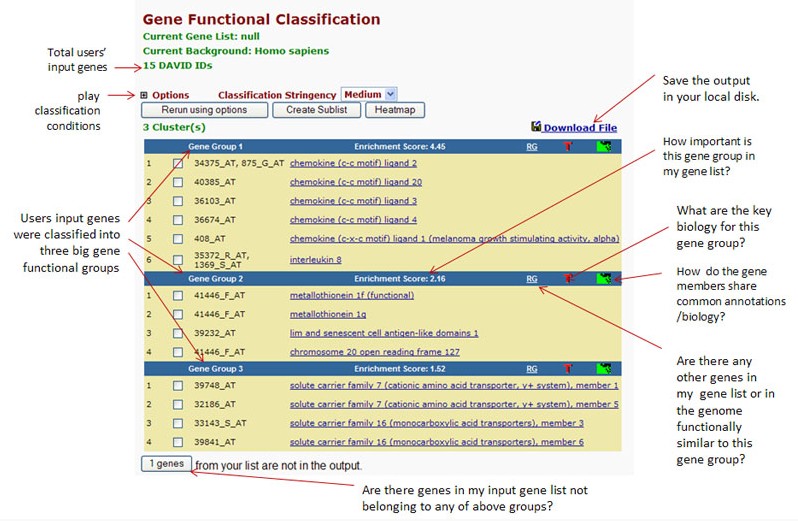
图2 Gene Functional Classification的分析结果

图3 2-D View展示gene-term关系
(3)Functional Annotation
该工具是DAVID最核心的分析内容,包含了三个子工具:
- Functional Annotation Chart
该工具提供gene-term的富集分析。相比于其他富集分析软件而言,DAVID在该功能上最显著的特点是,注释范围的可扩展性:从最初的GO注释,扩展到现在超过40中的注释种类,包括GO注释,KEGG注释,蛋白相互作用,蛋白功能区域,疾病相关,生物代谢通路,序列特点,异构体,基因功能总结,基因在组织里的表达和论文等。用户可以根据需要选择其中的某些或者所有种类的注释信息。
结果中以基因列表中富集的Terms为对象,将信息按照DAVID计算出来的p-value排列,同时链接指向更多的信息,见图4。
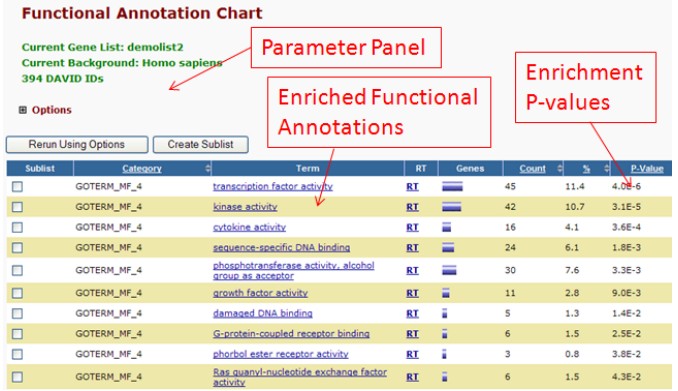
图4 Functional Annotation Chart的分析结果
- Functional Annotation Clustering
该工具使用类似于Gene Functional Classification工具的模糊聚类方法,基于注释共同出现的程度作聚类,对被注释上的Terms做聚类,即Terms被分成多组,并将给出聚类的分值。分值越高,代表该组内的基因在基因列表中越重要。同时还提供了2-D View,以热图形式展现聚类到同一组的基因和该组内各个Term之间的关系。
结果中(见图5),即被注释上的Terms作为聚类对象,用户可以根据聚类的分值找到重要的Terms。

图5 Functional Annotation Clustering的分析结果
- Functional Annotation Table
该工具实现了基因的功能注释,将输入列表中每个基因在选定数据库中的注释以表格形式呈现。结果见图6。.

图6 Functional Annotation Table的分析结果
(4)Gene ID Conversion
该工具实现不同数据库的基因标识间的转换。包含NCBI, PIR 和 Uniprot/SwissProt等重要数据库的基因标识信息。
结果如图7所示,左边的表格显示转换的情况,右边表格以列表呈现转换结果,和基因名称注释等。

图7 Gene ID Conversion分析结果
总结:
对于以上6项分析工具各有偏重点,下面给出一个指示图(见图8),帮助用户选择DAVID的各项分析工具。
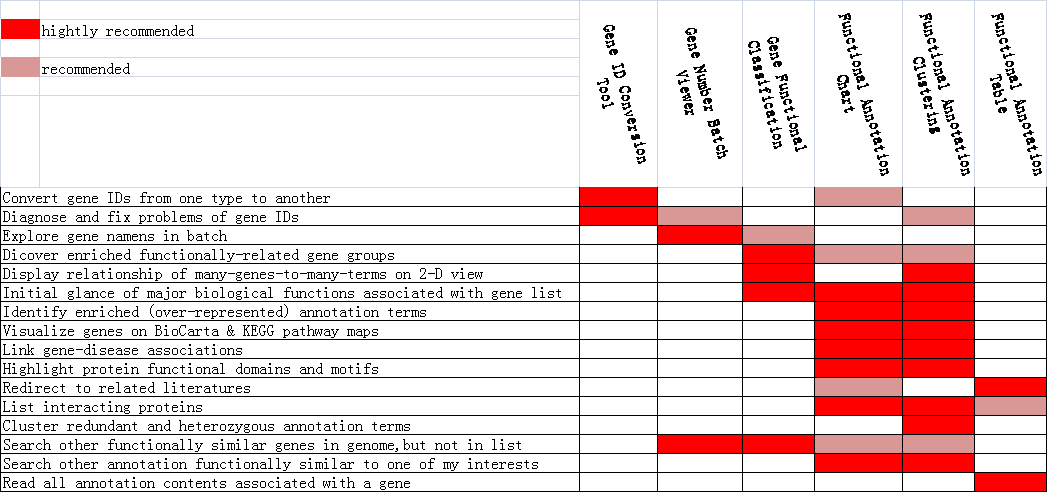
图8 DAVID各项分析工具的选择指示图
感谢@生物信息-小郑 在生物信息学QQ群(235461986)提供资料。
来自外部的引用