

尽管测序技术在不断发展,但不论是哪种测序平台,测序过程中都不可避免地存在一些错误。《Nature Review Genetics》近日发表了一篇很有意思的综述,谈论了NGS实验中错误的来源,以及如何利用重复来减少这些错误。文章的作者之一就是著名的遗传学家George Church。
在实验过程中使用重复是经过时间检验的法则,然而,由于成本较高,以及深度测序已提供一种类型的重复这一事实,很多实验室忽略了重复的重要性。但“读取深度”的范围有限,而其他类型的重复,如技术重复、生物学重复及跨平台的重复也非常有用。
2011年7月发表在《Nature Biotechnology》上的一篇文章就曾为研究人员以及杂志审稿人敲响了警钟。文章指出,测序技术并不能消除生物学差异。和芯片技术一样,新一代测序技术也需要生物学重复。
在测序过程中,由于我们能够对样品进行多重分析,故良好的实验设计能够利用技术重复,对同一个样品进行多次测序。目前一些研究使用技术重复,将数据集中起来用于进一步分析。
也有一些研究综合多个测序技术,来改善变异检出。例如,Illumina的短读长搭配PacBio的长读长,这就是个跨平台重复的很好例子。上个月,《PNAS》在线版上发表了一篇文章,就是将Illumina与PacBio的测序技术结合起来,来解决异构体鉴定和定量的问题。
生物学重复是指对同一条件下同一宿主的多个生物学样品进行测序,这也是差异表达分析的必需。这篇综述就详细介绍了利用不同组织的生物学重复来校正变异检出中的错误。
它深入研究了三个来自Complete Genomics的全基因组序列数据。作者将SNP归为一致或不一致,这取决于所有重复是否一致。利用多个打分方案,如读取深度、基因表达得分和基因组质量得分,作者利用ROC样的曲线分析了真阳性和真阴性的比例。有趣的是,相对于基因组质量得分和表达得分,以读取深度作为质量得分却表现不佳。
当然,光靠重复也不能解决所有的测序错误。对于参考基因组不完整、插入、缺失或重复的错误,我们还要另想办法解决。
此外,这篇文章还归类了错误的实验来源以及最早发现错误的文章。正如大家想象得一样,测序过程中的每一步都有可能混入错误,从样品制备到数据分析,因此大家一定要小心。
测序错误主要有三大类,分别来自样品制备、文库制备,以及测序和成像。
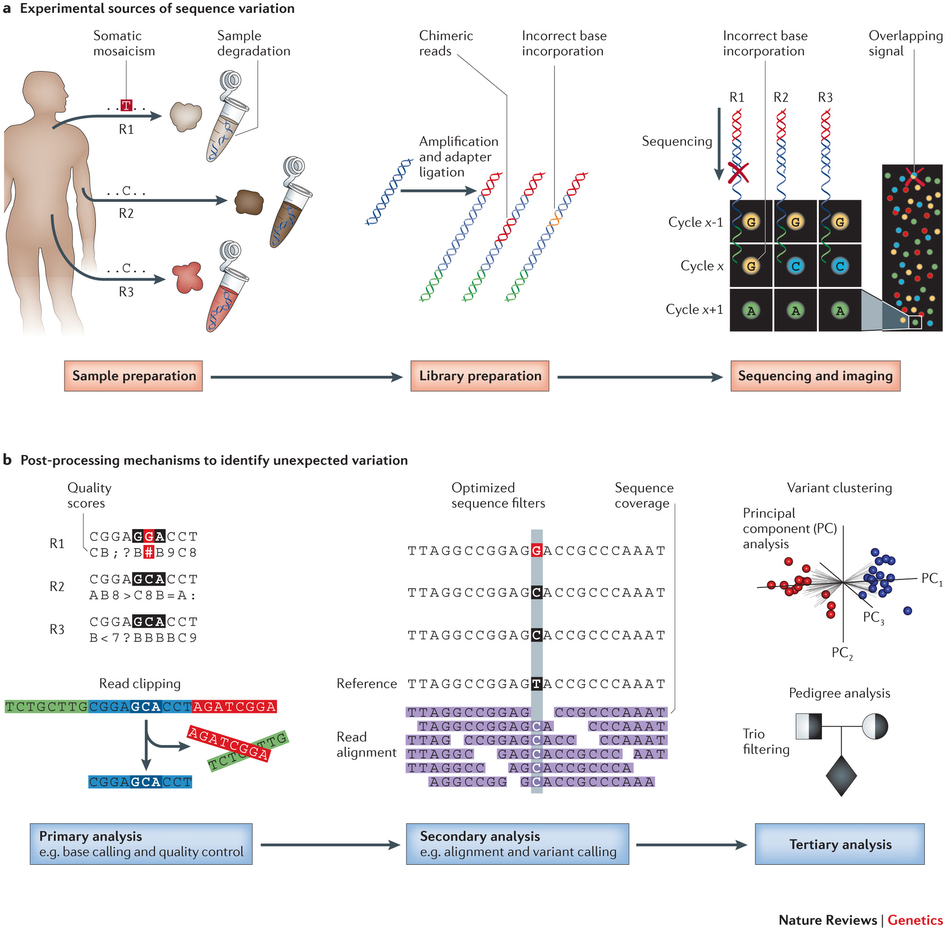
来源于样品制备的测序错误
1. 用户错误;例如,贴错标签。虽然这是个低级错误,但肯定不会没犯过。在芯片分析中,贴错标签和样品搞混可都是真事,有文献可查。
2. DNA或RNA的降解;例如,组织自溶,福尔马林固定石蜡包埋(FFPE)组织制备过程中的核酸降解和交联。
3. 异源序列的污染;例如,那些支原体和异种移植的宿主。
4. DNA起始量低。早在2005年人们就发现,在PCR过程中,DNA起始量低的模板会以序列依赖的方式产生虚假的突变,主要是从G转变为A。
来源于文库制备的测序错误
1. 用户错误;例如,一个样品的DNA残留到下一个,之前反应的污染。
2. PCR扩增错误。这个同上面第4点。
3. 引物偏向;例如,结合偏向,甲基化偏向,错配导致的偏向,非特异性结合和引物二聚体的形成,发夹结构和干扰环,熔解温度太高或太低引入的偏向。
4. 3’短捕获偏向,在高通量RNA测序的poly(A)富集过程中引入。
5. 独家突变;例如,那些由重复区域或独家变异的错配而引入的突变。
6. 机器故障;例如,PCR循环温度不正确。
7. 嵌合读取。
8. 条形码和/或接头错误;例如,接头污染,缺乏条形码多样性和不兼容的条形码。
来源于测序和成像的测序错误
1. 用户错误;例如,流动槽过载引起的簇crosstalk。
2. 移相;例如,不完整的延伸以及多个核苷酸而不是单个核苷酸的添加。
3. “Dead”荧光基团,受损的核苷酸以及重叠信号。
4. 序列背景;例如,富含GC,同源和低复杂度的区域,及均聚物。
5. 机器故障;例如,激光器、硬盘、软件和流体系统出故障。
6. 链的偏向。
由此可见,测序错误可能来源于实验的每一步。除了设计各种重复,大家在操作过程中也定要留心,避免让错误有空子可钻。
相关论文信息:
The role of replicates for error mitigation in next-generation sequencing
Kimberly Robasky, Nathan E. Lewis & George M. Church
Advances in next-generation sequencing (NGS) technologies have rapidly improved sequencing fidelity and substantially decreased sequencing error rates. However, given that there are billions of nucleotides in a human genome, even low experimental error rates yield many errors in variant calls. Erroneous variants can mimic true somatic and rare variants, thus requiring costly confirmatory experiments to minimize the number of false positives. Here, we discuss sources of experimental errors in NGS and how replicates can be used to abate such errors.
http://www.nature.com/nrg/journal/vaop/ncurrent/full/nrg3655.html
原文来自生物通,有部分修改。
1F
成本高,不可控因素也多,哎~
来自外部的引用