

首先,在开始之前我觉得有必要稍微科普缓冲一下,以便不使得不熟悉生物信息或基因组的客官们疑惑。
1.基因组:每个人都有一个基因组,这里的“基因组”并不只是“基因”的集合,基因是控制性状的遗传单元(什么是性状呢?性状也可以狭义的理解为个体的各种外在和内在特征,比如头发和眼睛颜色,高矮胖瘦,抵抗力强等),但是基因组所指的其实是我们的所有遗传信息,而不单单只是一些外在和内在特征,也包含很多目前而言不明其功能性(或者被认为无功能)的DNA序列。 其实说白了就是整一个的DNA序列!因而,基因也只是基因组的一个子集。此外,需要特别指出的是,我们虽都为“人”,但人与人之间的基因组是不一样的(即是多态的),彼此之间都存在着一些差异,即使是和父母或是兄弟姐妹之间去比较。这些差异也是造成我们彼此之间为何如此这般不同的一个重要原因。而这些差异也是基因组多态性的来源。
2.Reads:这里的reads是一个基因组测序(对测序原理感兴趣的客官请猛戳:三代基因组测序技术原理简介)中的名词,指的就是一段特定长度的DNA片段,这个长度取决于测序仪的读长。
3. 变异是一个相对的概念,只有在彼此的比较中才有存在的意义。目前关于人类基因组变异的讨论,都是以“人类基因组计划”中所组装出来的人类基因组作为参照物。以下谈到的涉及比对过程所用的基因组指的就是这个人类参考基因组。
4. 以下常出现“序列”,指的都是DNA序列片段。
OK!简单的科普就此完毕,剩余的在后面碰到了再说明,以下进入正文。
摘要:
人类基因组上的结构性变异研究对于基因组进化,群体多态性分析以及疾病易感性等方面的研究有着重要的意义。第二代短reads高通量测序技术的发展在带来了测序成本降低的同时,这种短读长的测序方式也给人类的变异检测带来了很大的挑战。这里我主要对当前常用的变异检测方法、软件以及他们各自的有确定做一个简要的小结。
人类基因组上的变异主要分为三大类:
1. 单核苷酸变异,(通常称为单核苷酸多态性,通俗的说法就是单个DNA碱基的不同,简称SNP);
2. 小的Indel(Insertion 和 Deletion的简),指的是在基因组的某个位置上所发生的小片段序列的插入或者删除,其长度通常在50bp以下(这个长度范围的变异可以利用Smith-Waterman 的比对算法来获得1,2);
3. 大的结构性变异,这种类型比较多,包括长度在50bp以上的长片段序列的插入或者删除、染色体倒位,染色体内部或染色体之间的序列易位,拷贝数变异,以及一些形式更为复杂的变异。
为了和SNP变异作区分,第2和第3类变异通常也被称为基因组结构性变异(Structural variation,简称SV)。这里值得一提的是,研究人员对基因组的结构性变异发生兴趣,主要是由于这几年的研究发现:
(1)虽然还未被广泛公认,但研究人员发现SV对基因组的影响比起SNP来说还要大3;
(2)基因组上的SV比起SNP而言,似乎更能用于解释人类群体多样性的特征;
(3)稀有且相同的一些结构性变异往往和疾病(包括一些癌症)的发生相关联甚至还是其致病的诱因4–6。
不过应该注意的地方是,大多数的结构性变异并不真正与疾病的发生相关联,但是却确实与周围环境的响应或者其他的一些表型多态性相联系。
近年来,随着芯片技术(这里的芯片技术和IT领域所说的芯不是同一个概念,这里指的是一种用于抓获基因组特定序列片段的技术)和第二代高通量测序技术的发展,人类基因组上的结构性变异图谱才被真正全面而又集中地进行了研究。生物信息研究人员已针对这两种不同的技术开发了许多相对应的软件用于检测基因组的结构性变异。相比较而言,虽然成本较高,但是基于测序的方法要明显优于芯片的检测,其中最重要的一个方面是,高通量测序技术能够在单碱基精度之下对全基因组范围内所有类型的变异进行检测,而芯片技术实际上只对大片段的序列删除比较敏感。
接下来我将会对目前基于第二代测序技术的变异检测方法进行介绍。
在各大生物信息学期刊(包括Nature,Science,Cell等这些顶级期刊)上都有许多关于介绍变异检测方面的文章。这里我大致说一下四篇自己觉得在这方面比较重要的文章:综述“Genome structural variation discovery and genotyping7”和综述“computational methods for discovering structural variation with next-generation sequencing”,这两篇文章所探讨的主要是,如何根据实验上和计算上的途径来检测和发现基因组上的各种变异,特别是对检测SVs而已。另外两篇文章则是基于千人基因组计划的,他们描述的是如何利用trio家系全基因组测序的数据和群体低覆盖度的数据来做变异检测的生物信息学方法8,9。然而需要指出的是,对于千人基因组计划,他们基本上只关注于一些大片段的序列删除和一些特定的序列插入方面的检测,而忽视了很多基因组上其他形式的变异。关于这方面的局限性,一方面可能是由于生物信息检测方法上的不完善,另一方面可能也和千人基因组本身的数据特点有关,使得他们难以准确地获得更多的信息。
目前主要有4种检测基因组上结构性变异的策略,分别为:(1)Read pair(也称为Pair-end Mapping,简称PEM);(2)Split read(简称SR);(3)Read Depth(简称RD)和(4)基于de novo组装的方法(图1)。同时生物信息研究人员也已开发了众多根据以上4中策略中一种或者多种的软件用于结构性变异的检测。接下来我将对这四种策略以及他们各自的特点逐一进行介绍。
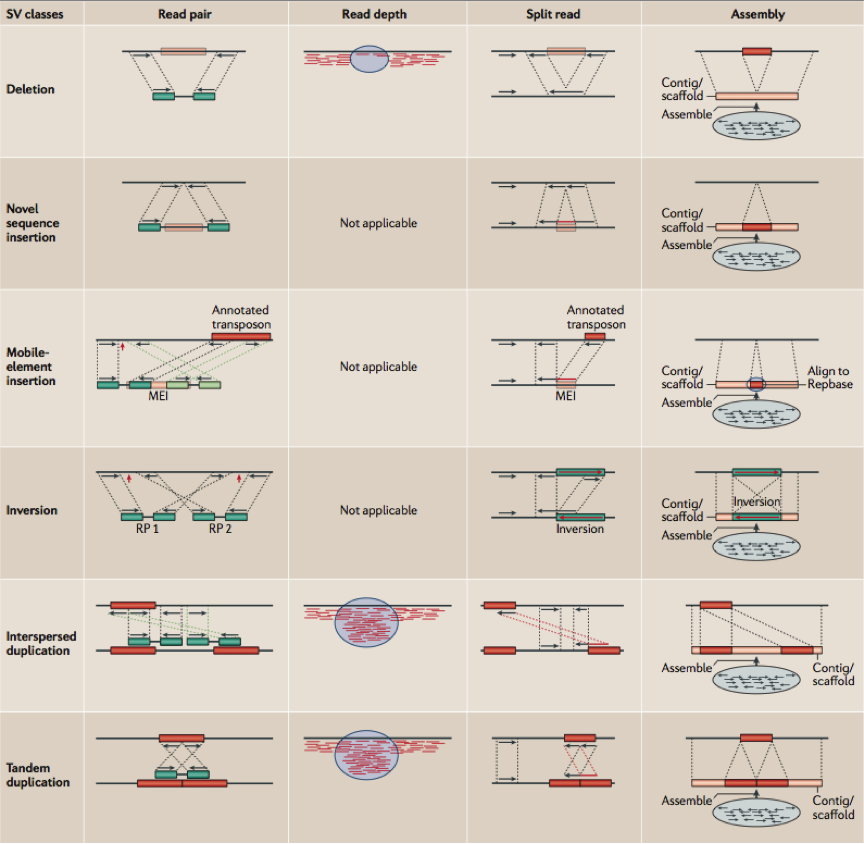
图1
1. 基于Pair-end Mapping(PEM)
图2是PEM方法的一个主要分析框架,理论上来讲,PEM方法能够检测到的变异类型包括:序列删除(deletion),序列插入(insertion),序列转置(inversion),染色体内部和染色体外部的易位(intra- and inter-chromosome translocation),序列串联倍增(tandem duplications)和序列在基因组上的散在倍增(interspersed duplications)。这里有两个地方需要指出,第一,对于序列删除的检测,其所能检测到的片段长度受插入片段长度的标准差(SD)所影响(这里的插入片段长度指的是测序之前在构建DNA测序文库阶段,所选取的经由超声波打断的DNA片段长度,这些片段也称之为测序片段,这是实验过程中的操作,并不是指基因组的变异),并且越大的序列删除约容易被检测到,并且准确性也越高;第二,其所能检测的序列插入,长度只能在插入片段长度的范围内,并且最大长度也受限于测序的插入片段长度的标准差。目前,Breakdancer是应用PEM方法的软件,也是在使用变异检测方面用得最广泛的软件之一。其他类似的软件还包括:VariationHunter10, Spanner, PEMer11等等。但是,事实上整个过程并不像流程图中看起来的那么简单,而且绝大多数的软件都在检测复杂的序列结构方面(如序列易位和序列倍增)存在很大的困难。
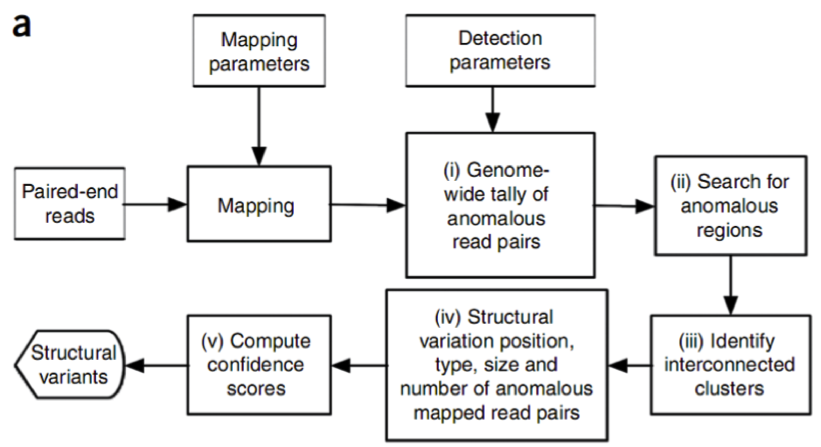
图2
2. Split Read(分裂read,简称SR)
对于这个方法,首先要求比对软件具备soft-clip reads的能力,如BWA 比对软件。我们知道目前illumina测序平台Pair-End测序的方法是对测序片段的两端来进行的,所以每次获得的都是来自同一个测序序列片段两端的一对read。当BWA成功地将这一对reads中的一条比对到参考序列上,而另一条却无法正常比上的时候,BWA会对这条read没能正常比上的read尝试在比对上的那条read附近使用更为宽松的Smith-Waterman局部比对策略搜索可能的比对位置。如果这条read只有一部分能够比上,那么BWA会对其进行soft-clip,而这里也往往是包含结构性变异的断点之处。Pindel12,这是目前唯一一个使用SR方法进行变异检测的软件。它在千人基因组计划和生物信息分析人员中被广泛使用。图1中也清楚地展示了Split reads的信号如何被用来进行结构性变异的检测。首先,在获得了单端唯一比对到基因组上的PE read之后,Pindel会将不能比上的那条read切开成2或者3小段,然后再分别重新按照用户所设置的最大序列删除长度去比对,并获得最终的比对位置和比对方向,而断点位置的确定则是根据soft-clipped的结果来获得。
Pindel 理论上能够检测所有长度范围内的deletion,和小片段的insertion(长度在50bp以下),inversion,tandem duplication和一些large insertion。不过目前,作者并未公开发布关于检测lager insertion的原理。Split-reads的一个优势就在于,它们精确到单碱基。但是也和大多数的PEM方法一样,Pindel同样无法解决复杂结构性变异的情形。
3. Read Depth (read 覆盖深度,简称RD)
目前存在两种利用Read depth的信息检测大拷贝数变异(Copy number variation,包括丢失序列和序列重复倍增,简称CNV)的策略。一种是,通过检测样本在一个参考基因组上read的深度分布情况来检测CNV,适用于单样本;另一种则是通过和识别出比较两个样本中所存在的丢失和重复倍增区,以此来获得相对的CNV,适用于case-control模型的样本。这有点像CGH芯片。CNVnator使用的是第一种策略,同时也广泛地被用于检测大的CNV。当然还有一些比较冷门的软件,但是由于他们没有发表相应的文章,这里就不再列举了。CNV-seq使用的是第二个策略。基于其原理,RD的方法能够很好地用于检测一些大的deletion或者duplication事件,但是对于小的变异事件就无能为力了。
4. 基于De novo assembly
理论上来讲,de novo assembly 的方法应该要算是基因组变异检测上最有效的方法了。就目前来说,它能够提供(特别是)对于long insertion和复杂结构性变异的最好检测方法。现在虽然研究人员开发了很多基于第二代测序技术数据来进行组装的软件,但是组装却仍然是一件棘手的事情,特别是脊椎动物的组装则更是如此。其中最主要的原因在于,脊椎动物基因组上所存在的重复性序列和序列的杂合会严重影响组装的质量,除去资金成本,这也在很大程度上阻碍了利用组装的方法在基因组变异检测方面的应用。
小结:
通过对上面四种不同的变异检测策略的比较可以发现,小长度范围内的变异以及较长的deletion,目前都能够较好地检测出来,但对于大多数的long insertion和更复杂的结构性变异情况,当前的检测软件基本都没法还解决。Assembly应是当前全面获得基因组上各种变异的最好方法,但是目前的局限却也发生在Assembly本身,若是基因组没能装得好,后面的变异检测就更是无从说起。从目前的情况看,de novo assembly的方法并不能很快进入实际的应用。因此,暂且不提assembly,其余的三种策略都各有各的优势,从目前的结果看,并没有哪一款软件能够一次性地将基因组上的各种不同情况变异类型都获得。因此就目前短reads高通量测序技术来说,最合适的方案应是结合多个不同的策略,将结果合并在一起,这样可以最大限度地将FP降低。HugeSeq pipeline13在这方面做了一个比较好的总结,这个软件整合了BreakDancer, CNVnator, Pindel,BreakSeq以及GATK的结果。能够给出一个相对比较准确的变异检测结果。最后这句怎么看起来像是在帮别人卖广告o(╯□╰)o。
参考文献:
1. DePristo, M. a et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics43, 491–8 (2011).
2. Albers, C. a et al. Dindel: accurate indel calls from short-read data. Genome research21, 961–73 (2011).
3. Conrad, D. F. et al. Europe PMC Funders Group Origins and functional impact of copy number variation in the human genome. 464, 704–712 (2012).
4. Campbell, P. J. et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nature genetics40, 722–9 (2008).
5. Berger, M. F. et al. The genomic complexity of primary human prostate cancer. Nature470, 214–20 (2011).
6. Stephens, P. J. et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell144, 27–40 (2011).
7. Alkan, C., Coe, B. P. & Eichler, E. E. Genome structural variation discovery and genotyping. Nature reviews. Genetics12, 363–76 (2011).
8. Mills, R. E. et al. Mapping copy number variation by population-scale genome sequencing. Nature470, 59–65 (2011).
9. Africa, W. A map of human genome variation from population-scale sequencing. Nature467, 1061–73 (2010).
10. Hormozdiari, F., Alkan, C., Eichler, E. E. & Sahinalp, S. C. Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome research19, 1270–8 (2009).
11.Korbel, J. O. et al. PEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome biology10, R23 (2009).
12.Ye, K., Schulz, M. H., Long, Q., Apweiler, R. & Ning, Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics (Oxford, England)25, 2865–71 (2009).
13.Lam, H. Y. K. et al. Detecting and annotating genetic variations using the HugeSeq pipeline. Nature biotechnology30, 226–9 (2012).
原文来自:http://www.cnblogs.com/huangshujia/p/3344911.html
1F
安不上breakdancer 怎么办?跪求大神指点!!!