

1. Variant Calling
GATK在这一步里面提供了两个工具进行变异检测——UnifiedGenotyper和HaplotypeCaller。其中HaplotypeCaller一直还在开发之中,包括生成的结果以及计算模型和命令行参数一直在变动,因此,目前使用比较多的还是UnifiedGenotyper。此外,HaplotypeCaller不支持Reduce之后的bam文件,因此,当选择使用HaplotypeCaller进行变异检测时,不需要进行Reduce reads。
UnifiedGenotyper是集合多种变异检测方法而成的一种Variants Caller,既可以用于单个样本的变异检测,也可以用于群体的变异检测。UnifiedGenotyper使用贝叶斯最大似然模型,同时估计基因型和基因频率,最后对每一个样本的每一个变异位点和基因型都会给出一个精确的后验概率。
e.g.
java -jar GenomeAnalysisTK.jar
-glm BOTH
-l INFO
-R hg19.fa
-T UnifiedGenotyper
-I ChrALL.100.sam.recal.08-3.grp.reduce.bam
-D dbsnp_137.hg19.vcf
-o ChrALL.100.sam.recal.10.vcf
-metrics ChrALL.100.sam.recal.10.metrics
-stand_call_conf 10
-stand_emit_conf 30
上述命令将对输入的bam文件中的所有样本进行变异检测,最后生成一个vcf文件,vcf文件中会包含所有样本的变异位点和基因型信息。但是现在所得到的结果是最原始的、没有经过任何过滤和校正的Variants集合。这一步产生的变异位点会有很高的假阳性,尤其是indel,因此,必须要进行进一步的筛选过滤。这一步还可以指定对基因组的某一区域进行变异检测,只需要增加一个参数 -L:target_interval.list,格式是bed格式文件。
主要参数解释:
-A: 指定一个或者多个注释信息,最后输出到vcf文件中。
-XA: 指定不做哪些注释,最后不会输出到vcf文件中。
-D: 已知的snp文件。
-glm: 选择检测变异的类型。SNP表示只进行snp检测;INDEL表示只对indel进行检测;BOTH表示同时检测snp和indel。默认值是SNP。
-hets: 杂合度的值,用于计算先验概率。默认值是0.001。
-maxAltAlleles: 容许存在的最大alt allele的数目,默认值是6。这个参数要特别注意,不要轻易修改默认值,程序设置的默认值几乎可以满足所有的分析,如果修改了可能会导致程序无法运行。
-mbq: 变异检测时,碱基的最小质量值。如果小于这个值,将不会对其进行变异检测。这个参数不适用于indel检测,默认值是17。
-minIndelCnt: 在做indel calling的时候,支持一个indel的最少read数量。也就是说,如果同时有多少条reads同时支持一个候选indel时,软件才开始进行indel calling。降低这个值可以增加indel calling的敏感度,但是会增加耗费的时间和假阳性。
-minIndelFrac: 在做indel calling的时候,支持一个indel的reads数量占比对到该indel位置的所有reads数量的百分比。也就是说,只有同时满足-minIndelCnt和-minIndelFrac两个参数条件时,才会进行indel calling。
-onlyEmitSamples:当指定这个参数时,只有指定的样本的变异检测结果会输出到vcf文件中。
-stand_emit_conf:在变异检测过程中,所容许的最小质量值。只有大于等于这个设定值的变异位点会被输出到结果中。
-stand_call_conf:在变异检测过程中,用于区分低质量变异位点和高质量变异位点的阈值。只有质量值高于这个阈值的位点才会被视为高质量的。低于这个质量值的变异位点会在输出结果中标注LowQual。在千人基因组计划第二阶段的变异检测时,利用35x的数据进行snp calling的时候,当设置成50时,有大概10%的假阳性。
-dcov: 这个参数用于控制检测变异数据的coverage(X),4X的数据可以设置为40,大于30X的数据可以设置为200。
注意:GATK进行变异检测的时候,是按照染色体排序顺序进行的(先call chr1,然后chr2,然后chr3…最后chrY),并非多条染色体并行检测的,因此,如果数据量比较大的话,建议分染色体分别进行,对性染色体的变异检测可以同常染色体方法。
大多数参数的默认值可以满足大多数研究的需求,因此,在做变异检测过程中,如果对参数意义不是很明确,不建议修改。
2. 对原始变异检测结果进行过滤(hard filter and VQSR)
这一步的目的就是对上一步call出来的变异位点进行过滤,去掉不可信的位点。这一步可以有两种方法,一种是通过GATK的VariantFiltration,另一种是通过GATK的VQSR(变异位点质量值重新校正)进行过滤。
通过GATK网站上提供的最佳方案可以看出,GATK是推荐使用VASR的,但使用VQSR数据量一定要达到要求,数据量太小无法使用高斯模型。还有,在使用VAQR时,indel和snp要分别进行。
VQSR原理介绍:
这个模型是根据已有的真实变异位点(人类基因组一般使用HapMap3中的位点,以及这些位点在Omni 2.5M SNP芯片中出现的多态位点)来训练,最后得到一个训练好的能够很好的评估真伪的错误评估模型,可以叫他适应性错误评估模型。这个适应性的错误评估模型可以应用到call出来的原始变异集合中已知的变异位点和新发现的变异位点,进而去评估每一个变异位点发生错误的概率,最终会给出一个得分。这个得分最后会被写入vcf文件的INFO信息里,叫做VQSLOD,就是在训练好的混合高斯模型下,一个位点是真实的概率比上这个位点可能是假阳性的概率的log odds ratio(对数差异比),因此,可以定性的认为,这个值越大就越好。
VQSR主要分两个步骤,这两个步骤会使用两个不同的工具:VariantRecalibrator和ApplyRecalibration。
i) VariantRecalibrator
VariantRecalibrator:通过大量的高质量的已知变异集合的各个注释(包括很多种,后面介绍)的值来创建一个高斯混合模型,然后用于评估所有的变异位点。这个文件最后将生成一个recalibration文件。
原理简单介绍: 这个模型首先要拿到真实变异数据集和上一步骤中得到的原始变异数据集的交集,然后对这些SNP值相对于具体注释信息的分布情况进行模拟,将这些变异位点进行聚类,最后根据聚类结果赋予所有变异位点相应的VQSLOD值。越接近聚类核心的变异位点得到的VQSLOD值越高。
ii) ApplyRecalibration
ApplyRecalibration:这一步将模型的各个参数应用于原始vcf文件中的每一个变异位点,这时,每一个变异位点的注释信息列中都会出现一个VQSLOD值,然后模型会根据这个值对变异位点进行过滤,过滤后的信息会写在vcf文件的filter一列中。
原理简单介绍: 在VariantRecalibrator这一步中,每个变异位点已经得到了一个VQSLOD值了,同时,这些LOD值在训练集里也进行了排序。当你在这一步中设置一个tranche sensitivity 的阈值(这个阈值一般是一个百分数,如设置成99%),那么,如果LOD值从大到小排序的话,这个程序就会认为在这个训练集中,LOD值在前99%的是可信的,当这个值低于这个阈值,就认为是错误的。最后,程序就会用这个标准来过滤上一步call出来的原始变异集合。如果LOD值超过这个阈值,在filter那一列就会显示PASS,如果低于这个值就会被过滤掉,但是这些位点仍然会显示在结果里面,只不过会在filter那一列标示出他所属于的tranche sensitivity 的名称。在设置tranche sensitivity 的阈值时,要兼顾敏感度和质量值。
对高斯混合模型生成图片的解释:
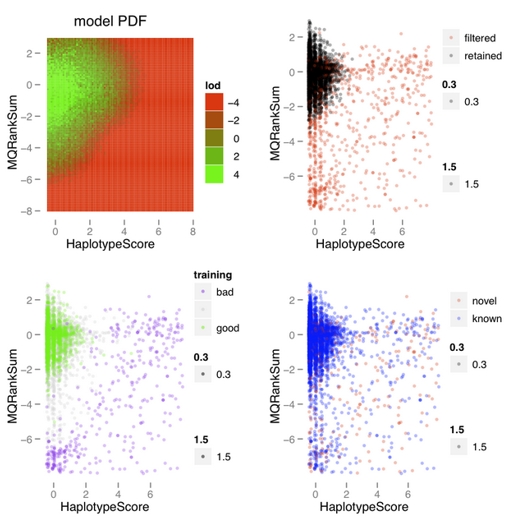
在VariantRecalibrator这一步,程序会通过已知位点来训练概率模型,训练完成后会生成一组图片,而且每对注释信息都对应一组图片(上图),这组图片能够帮助我们理解一个概率模型是否与我们的数据相匹配,也就是说这个模型能不能很好的区分假阳性和真实位点。
上图是第一步完成后生成的一个报告的一部分,图中只表示了一对注释所对应的图。左上角的图表示的是适合当前数据的概率密度图,绿色区域表示高质量变异位点所在位置,红色区域表示低质量概率分布区域。如果变异位点分布在红色区域,则会被过滤掉。右上角图中红色的点表示在经过VQSR之后被过滤掉的变异位点,黑色的表示的是留下来的。红色的表示的都是没有达到所设定的tranche sensitivity 阈值的点。左下角的图表示的是用来训练模型的点,绿色的点表示通过训练进入到ApplyRecalibration的变异位点,紫色的点则表示质量值很低的,没有达到质量要求的点。右下角的图表示的是已知的和新发现的变异位点的分布,红色的点表示新发现的变异位点,而蓝色的点表示的是已知的变异位点,看这幅图就是看这两个注释信息能不能很好的区分已知的点(大部分是真实的)和未知的点(大部分是假阳性)。
从图中可以看出,这个模拟结果可以很好的将真实的变异位点和假阳性变异位点分开(左下图),形成了明显的界限,也就是说,如果一个变异位点的这两个注释值,只要有一个落在了界限之外,就会被过滤掉。最主要的是要看右边两个图片,只要能很好的区分开novel和known以及filtered和retained就可以。其实在如何选择注释值存在一定得主观性,因此,在做VariantRecalibrator时可以做两次,第一次尽可能的多的选择这些注释值,第一遍跑完之后,选择几个区分好的,再做一次VariantRecalibrator,然后再做ApplyRecalibration。具体每个注释值得意义可以参考:
http://www.broadinstitute.org/gatk/guide/tagged?tag=annotation这个网址中的内容,有每个注释的详细信息的链接。
tranche值的设定
前面提到了,这个值得设定是用来在后续的ApplyRecalibration中如何根据这个阈值来过滤变异位点的,也就是说,如果这个值设定的比较高的话,那么最后留下来的变异位点就会多,但同时假阳性的位点也会相应增加;如果设定的低的话,虽然假阳性会减少,但是会丢失很多真实的位点。因此,跟选择注释时一样,可以run两遍VariantRecalibrator,第一遍的时候多写几个阈值,第一遍跑完之后看结果,看那个阈值好,选择一个最好的阈值,再run一遍VariantRecalibrator。至于说怎么区分好坏,有几个标准:
1. 看结果中已知变异位点与新发现变异位点之间的比例,这个比例不要太大,因为大多数新发现的变异都是假阳性,如果太多的话,可能假阳性的比例就比较大;
2. 看保留的变异数目,这个就要根据具体的需求进行选择了。
3. 看TI/TV值,对于人类全基因组,这个值应该在2.15左右,对于外显子组,这个值应该在3.2左右,不要太小或太大,越接近这个数值越好,这个值如果太小,说明可能存在比较多的假阳性。
千人中所选择的tranche值是99,仅供参考。
注意:Indel不支持tranche值的选择,另外,一部分注释类型在做indel的校正时也不支持,具体信息可以详查GATK网站。
当数据量太小时,可能高斯模型不会运行,因为变异位点数满足不了模型的统计需求。这时候可以通过降低--maxGaussian的值,让程序运行。这个值表示的是程序将变异位点分成的最大的组数,降低这个值让程序把变异位点聚类到更少的组里面,使每个组中的变异位点数增加来满足统计需求,但是这样做降低程序分辨真伪的能力。因此,在运行程序的时候,要对各方面进行权衡。
补充:
原文来自:http://blog.sina.com.cn/s/blog_12d5e3d3c0101qu6r.html
1F
你好,请问使用UnifiedGenotyper同时对多个样本进行snp calling 时,这些样本必须是同类型的吗?比如肿瘤样本和正常样本可以放在一起进行snp calling 吗?如果可以放在一起,怎么找肿瘤样本中特异的snp呢?谢谢。
2F
这里的数据可以是cleandata吗?
来自外部的引用