

有趣的是,大部分的short read比对工具都是由中国人写出来的。因此可以说华大基因(BGI, Beijing Genomics Institute, Chinese Academy of Science)是中国NGS测序技术的摇篮。
速度上较有优势的short read(短序)比对工具最早出现的是SOAP(表1)。它很好地解决了一个问题,那就是如何在小内存(4G)的机器上将短序比对至人类基因组这样的大数据上去。我们都知道,人类基因组的大小为3.2G(表2),光把这样大的数据读入内存都是一件不太容易的事情。所以SOAP对NGS的贡献是值得我们记住的。SOAP在设计之初是针对single-end reads, 所以对paired-end的支持不被大家看好。它的成功也逐步被后起之秀所掩盖。
表1:发表时间
| 软件 | 发表年代 | PMID |
| SOAP | 2008 | 18227114 |
| Maq | 2008 | 18714091 |
| BWA | 2009 | 19451168 |
| Bowtie | 2009 | 19261174 |
| NovoAlign | 2009 | |
| Subread | 2013 | 23558742 |
表2:基因组大小(数据来源Ensenbl, release 72)
| 物种 | 碱基数 | 基因数 |
| 人 homo sapiens | 3.3G | ~21K |
| 小鼠 mus musculus | 3.5G | ~23K |
| 大鼠 rattus norvegicus | 2.6G | ~23K |
| 果蝇 Drosophila melanogaster | 169M | ~14K |
| 线虫 Caenorhabditis elegans | 103M | ~21K |
之后先后出现了两个重要的比对软件,MAQ/BWA以及Bowtie。MAQ/BWA是Heng Li发表的。Li是从华大基因走出来的人,后来去了Wellcome Trust Sanger Institute, 现在在哈佛Broad Institute。MAQ的引用率非常高,并成为了Li的成名作。之后写作的BWA以准确率高而闻名,是SNP分析的首选比对软件。
而Bowtie借着其算法上的优势,在运算速度上一举成名。如果对速度的要求高于准确率的时候,bowtie就成了不二选择。bowtie被广泛地应用于ChIP-seq, RNA-seq的分析当中。
NovoAlign是一款商业软件,但是如果只是科研用途的话,可以直接从其网站上下载到编译好的程序(只支持unix/linux/mac)。它也有MPI版本。但是因为在单机上运行效率问题以及商业化的原因,它的应用并不象BWA和Bowtie那样广泛。
Subread是最新出现的比对软件。作者Wei Shi在文章中声称subread在速度和准确率上都较之前的主流软件有优势。并且它还有R/Bioconductor版本。但是在SEQanswers的讨论中,他与BWA的作者Heng Li打起了口水战,他们都声称自己的软件才是准确率最高的。甚至两人还给出了截然相反的两组比较数据(下图)
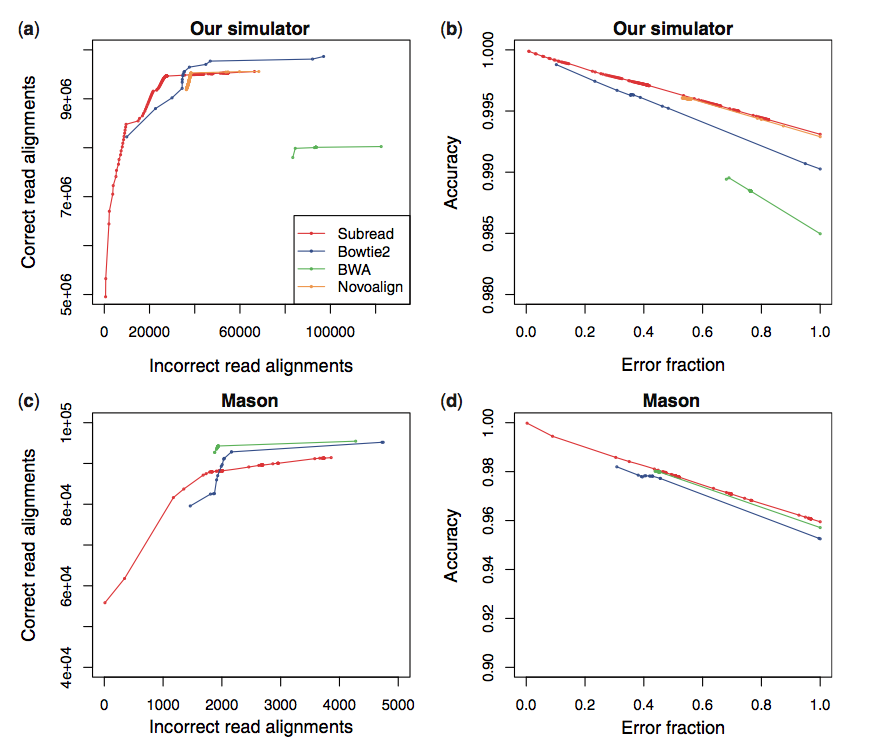
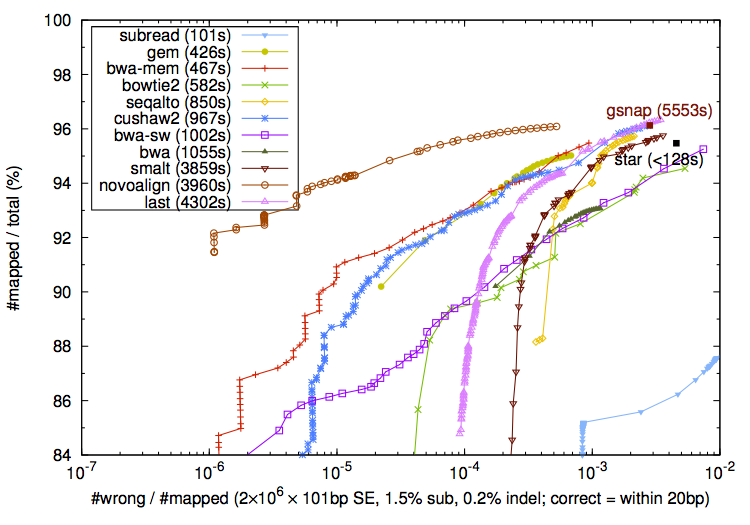
由于公说公有理,婆说婆有理,加上subread还需要时间来考验,所以现在还无从判断谁的更具有优势。
由于选择变多了,人们往往会变得无所适从。就我个人经验而言(其实是对研究机构前人脚本学习和接受),对于ChIP-seq, RNA-seq,多使用bowtie2,因为它快速,下游结合cufflinks等结果验证率很高。对于SNP, Indels, methylation分析,使用BWA,下游结合GATK可能会好一点。
原文来自:http://pgfe.umassmed.edu/ou/archives/3197