

本文将以MACS为例,介绍ChIP-seq数据的处理流程。为节省篇幅,本文略去测序数据预处理、mapping reads等步骤,直接从peak-calling开始讲起。
一、首先粗略地介绍一下MACS的基本原理。
TF在基因组上的结合其实是一个随机过程,基因组的每个位置其实都有机会结合某个TF,只是概率不一样,说白了,peak出现的位置,是TF结合的热点,而peak-calling就是为了找到这些热点。
如何定义热点呢?通俗地讲,热点是这样一些位置,这些位置多次被测得的read所覆盖(我们测的是一个细胞群体,read出现次数多,说明该位置被TF结合的几率大)。那么,read数达到多少才叫多?这就要用到统计检验喽。假设TF在基因组上的分布是没有任何规律的,那么,测序得到的read在基因组上的分布也必然是随机的,某个碱基上覆盖的read的数目应该服从二项分布。这其实和高中大学课本上抽小球的过程是类似的。当n很大,p很小时,二项分布可以近似用泊松分布替代,在这里:
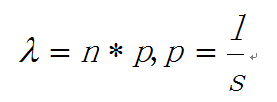
是泊松分布唯一的参数,n是测序得到的read总数目,l是单个read的长度,s是基因组的大小。有了分布,我们可以算出在某个置信概率(如0.00001)下,随机情况下,某个碱基上可以覆盖的read的数目的最小值,当实际观察到的read数目超过这个值(单侧检验)时,我们认为该碱基是TF的一个结合热点。反过来,针对每一个read数目,我们也可以算出对应的置信概率P。
但是,这只是一个简化的模型,实际情况要复杂好多。比如,由于测序、mapping过程内在的偏好性,以及不同染色质间的差异性,相比全基因组,某些碱基可能内在地会被更多的read所覆盖,这种情况得到的很多peak可能都是假的。MACS考虑到了这一点,当对某个碱基进行假设检验时,MACS只考虑该碱基附近的染色质区段(如10k),此时,上述公式中n表示附近10k区间内的read数目,s被置为10k。当有对照组实验(Control,相比实验组,没有用抗体捕获TF,或用了一个通用抗体)存在时,利用Control组的数据构建泊松分布,当没有Control时,利用实验组,稍大一点的局部区间(比如50k)的数据构建泊松分布。
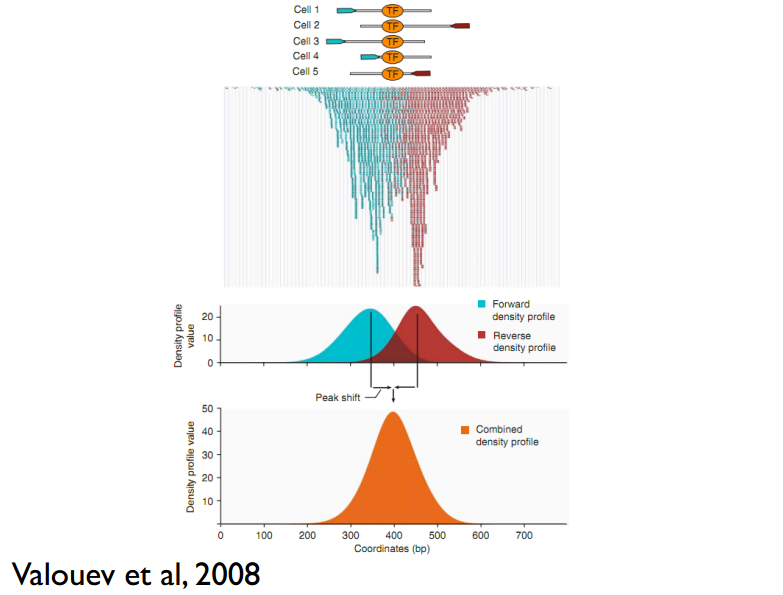
这儿还有一个问题,read只是跟随着TF一起沉淀下来的DNA fragment的末端,read的位置并不是真实的TF结合的位置。所以在peak-calling之前,延伸read是必须的。不同TF大小不一样,对read延伸的长度也理应不同。我们知道,测得的read最终其实会近似地平均分配到正负链上,这样,对于一个TF结合热点而言,read在附近正负链上会近似地形成“双峰”。MACS会以某个window size扫描基因组,统计每个window里面read的富集程度,然后抽取(比如1000个)合适的(read富集程度适中,过少,无法建立模型,过大,可能反映的只是某种偏好性)window作样本,建立“双峰模型”。最后,两个峰之间的距离就被认为是TF的长度D,每个read将延伸D/2的长度。见下图:
当有对照组实验存在时,MACS会进行两次peak calling。第一次以实验组(Treatment)为实验组,对照组为对照组,第二次颠倒,以实验组为对照组,对照组为实验组。之后,MACS对每一个P计算了相应的FDR(False Discovery Rate)值:
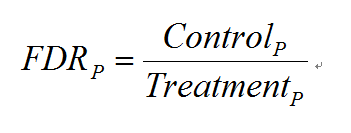
表示第二次peak calling(颠倒的)得到的置信概率小于P的peak的个数。表示第一次peak calling得到的置信概率小于P的peak的个数。FDR综合利用了实验组和对照组的信息,显然,FDR越小越好。
二、MACS peak-calling pipeline:
MACS有两个版本,分别是MACS14和MACS2。MACS2在很多方面都对MACS14做了重大改进,但目前还在测试阶段。我们依然以MACS14为例进行说明。
安装下载源代码:http://github.com/downloads/taoliu/MACS/MACS-1.4.2-1.tar.gz
- tar xvzf MACS-1.4.2-1.tar.gz
- cd MACS-1.4.2
- python setup.py install –prefix /your_directory/
–prefix用于指定安装目录
最后,修改环境变量:
- export PATH = /your_directory/bin:$PATH
- export PYTHONPATH = /your_directory/lib/python2.X/site-packages/:$PYTHONPATH
更多细节见安装包内的INSTALL文件。
假设我们现在有mouse的一组CTCF的ChIP-seq测序数据CTCF.fastq,首先,我们把这些reads map到mouse基因组(这里我们采用mm10)上。假设基因组的index文件已经建好,存在/path_to/文件夹下。
- bowtie –m 1 -S -q /path_to/mm10 CTCF.fastq CTCF.sam
-m 最终只保留map上一次的reads
-S 输出文件格式是SAM
-q 输入文件格式是fastq
peak-callingmacs14 -t CTCF.sam -n CTCF –g mm-t 实验组数据文件名(相对对照组control而言,后面会进一步说明)-n 输出文件名前缀
-g 基因组的大致大小,-g number。MACS内置了一些基因组长度,“mm”表示小鼠的,“hs”表示人的,“ce”表示线虫,“dm”是果蝇。
运行成功后,将得到如下文件:
CTCF_model.r,CTCF_peaks.bed,CTCF_peaks.xls,CTCF_summits.bed
其中,CTCF_model.r以代码的形式保存了“双峰模型”。在终端中输入:
- Rscript CTCF_model.r
得到名为CTCF_model.pdf的文件,打开,如下图所示:
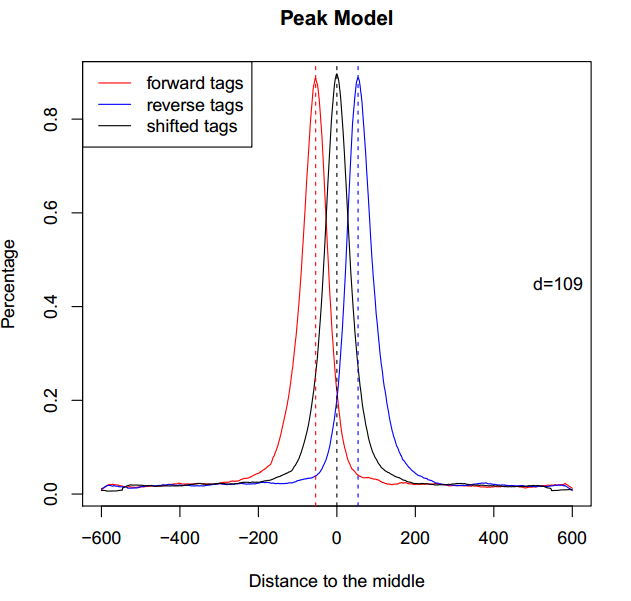
在某些情况下,比如,对组蛋白修饰的ChIP-seq数据peak-calling时,“双峰模型”会建立失败,这是因为组蛋白修饰往往并不是孤立存在的,可能很长一段染色质区间都被同一个组蛋白修饰占据,换句话说,组蛋白修饰的peak并不典型。这时,只要多加一个参数:
–nomodel –shiftsize=number
–nomodel将略过“双峰模型”建立的过程,而–shiftsize将人为指定reads延伸的长度。因为我们知道一个核小体上大概缠绕着147bp长的DNA,在对组蛋白修饰做peak-calling时我们可以指定:
–nomodel –shiftsize=73
CTCF_peaks.bed详细列出了每一个peak的位置信息和可信度(最后一列:),BED文件格式详见:http://genome.ucsc.edu/FAQ/FAQformat.html#format1
CTCF_peaks.xls相比CTCF_peaks.bed包含了更多信息,包括本次程序运行的详细参数,每个peak的summit(覆盖最多延伸后reads的那个位置)信息等。
当我们有对照组实验数据时(Control.sam):macs14 -t CTCF.sam -c Control.sam -n CTCF –g mm输出文件类似,不过多出一个CTCF_negative_peaks.xls,顾名思义,是互换对照组实验组后得到的peak。
其他常用参数:-bw number 建立“双峰模型”过程中window size的一半,默认300bp.-p Pvalue 设定peak置信概率的临界值(threshold),默认0.00001,对于H3k36me3、H3k27me3、H3k9me3等具有“非常规”特征的peak(broad peak)而言,此参数可以稍微设大一点,比如0.001。
-m number1,number2 建立“双峰模型”用到,设定挑选的window上reads的富集程度(fold enrichment,相对全基因组而言),默认10,30。
-slocal=number -llocal=number 共同确定MACS动态计算时所考察的局部区间的长度。默认参数,-slocal=1000 -llocal=10000。这儿多说几句,除了建立“双峰模型”,在寻找peak过程中,MACS依然会以2倍于-bw的window扫描基因组,对于当前window而言(-slocal,-llocal取默认参数):
其中,是基于全基因组的(实验组),基于当前window(默认参数下600bp)。当Control组存在时,均是基于Control组数据估算,当Control组不存在时,将不考虑,只考虑。
-w/-B 建立wig或BED格式的raw signal(最高精确到每个碱基上reads的覆盖情况)文件,默认每条染色体建一个。
-S 只建立一个raw signal文件。
-space 与-w搭配使用,确定wig文件的分辨率,默认10bp。
原文来自:http://www.zilhua.com/1562.html
来自外部的引用