

1. ICORN 简介
ICORN (Iterative Correction Of Reference Nucleotides), 能通过将 reads 比对到基因组,从而修正 SNP 和 小于 3bp 的 INDEL 位点。现在出了新的版本 ICORN2 。
ICORN 官网: http://icorn.sourceforge.net/
ICORN 参考文献: Otto T D, Sanders M, Berriman M, et al. Iterative Correction of Reference Nucleotides (iCORN) using second generation sequencing technology[J]. Bioinformatics, 2010, 26(14): 1704-1707.
2. ICORN 原理
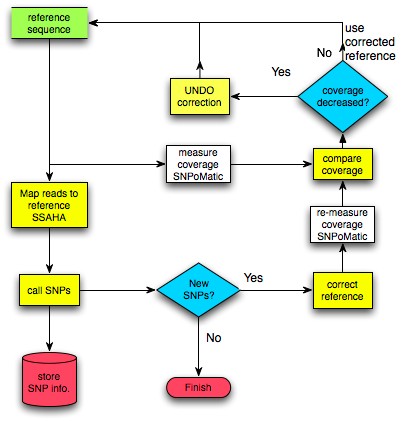
ICORN 的原理如下图所示。ICORN2 调用 SMALT 将 reads map 到基因组序列上;然后 Call SNPs 和 <=3bp INDELs;根据其位点的覆盖度情况决定基因组上该位点的核苷酸类型。最后通过多轮迭代直到不能 call 到新的 variants 为止。
3. ICORN 的下载和安装
ICORN 的使用会调用第三方的软件: GATK、SMALT、samtools 和 SNP-o-AMTIC 。
- $ wget ftp://ftp.sanger.ac.uk/pub4/resources/software/pagit/ICORN2/icorn2.V0.95.tgz
- $ tar zxf icorn2.V0.95.tgz -C /opt/biosoft/
- $ /opt/biosoft/ICORN2/icorn2.sh --help
4. ICORN 的使用
4.1 使用前准备
准备的输入文件是: readroot_1.fastq、readroot_2.fastq、genome.fasta,将这3个文件放置于工作目录中。
同时,需要给一些环境变量赋值:
设定程序所在的目录
- $ export ICORN2_HOME=/opt/biosoft/ICORN2/
设定运行的线程数
- $ export ICORN2_THREADS=24
设定输出信息的多或少,对 debug 有用
- $ export ICORN2_VERBOSE=2
4.2 运行软件
- $ /opt/biosoft/ICORN2/icorn2.sh readroot 350 genome.fasta 1 3
以上命令第 2 个参数是数据的 insert size; 1 和 3 代表迭代的起始和终止,表示迭代 3 次。如果需要继续迭代,则设置起始为 4 。上述程序的结果文件为 ICORN2.Query.contigs.fa.4 。在 ICORN2_3 文件夹中也含有该 fasta 文件,同时程序也生成了一个 gff 文件。作者推荐将这两个文件载入到 artemis 基因组浏览器中进行查看。
5. 思考
有些人在有参考基因组的状况下做了重测序,总是想要得到其重测序的基因组结果。可以使用 ICORN 对参考基因组进行修正,即得到了对应其品种的基因组序列。
原文来自:http://www.chenlianfu.com/?p=2146