

刚接触高通量测序的同学,可能会接触到barcode index等名词。
为什么要有barcodes ?
原因很简单,一个lane有效数据30来G
我们并常常不可能拿一个样品测那么大数据量,尤其是RNA-seq,哈哈,
所以在建库的时候,对每一个样品接头上加上不同的标签序列,然后用于测序,在测序结果中,依据之前标签与样品的对应关系,就可以获得对应样品的数据,从而就有所谓的数据量为1G,2G。。。等等
本文将同时大概介绍下测序原理,方便同学们更好的了解测序数据的特点
下图来自文献《Multiplexed Illumina sequencing libraries from picogram quantities of DNA》
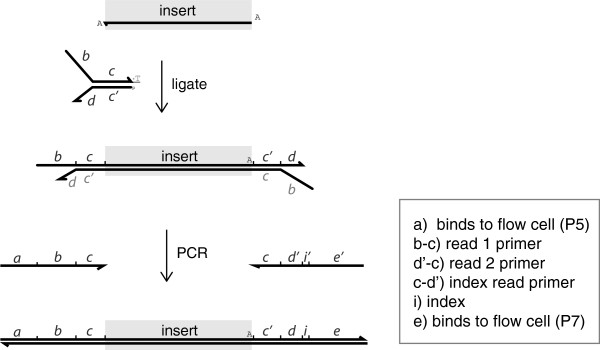
对于illumina的hiseq平台而言,测序前,我们需要建库。
1. 建库的第一步即对mRNA片段化,
2. 随后是片段大小筛选,一般RNA-seq取200bp,DNA-seq取500bp(重测序),即为图中insert size,也常称为fragment size(修正,一般fragment size还包括引物序列长度),中文常称为插入片段大小
3. 加正向接头,接头上(部分<3端>与正向引物完全相同的序列),加反向接头
4. cot扩展,即PCR扩增
5. 测序
###关于测序一步,此处要重点讲下
明确一点,barcode在反向接头中
正向接头(包含正向引物序列)+插入片段+反向接头(包含反向引物序列+index/barcodes+…)
测序时非常简单,以PE100为例
正向引物读100bp
反向引物读100bp
barcode引物读barcode长度的bp数
这里设计到一个接头污染,引物污染的问题,具体请自行思考
原文来自:http://www.pineapplechina.com/?p=63
1F
wow,我终于能看懂了。
2F
能不能给点正向引物序列和其对应的反向引物序列,对照看看~