

1:分析流程图如下
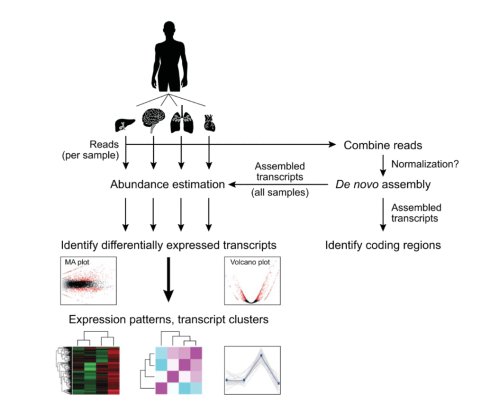
2: 首先就是将样本的reads合并在一起命令如下:
- cat 1M_READS_sample/*.left.fq > reads.ALL.left.fq
- cat 1M_READS_sample/*.right.fq > reads.ALL.right.fq
3:开始拼接
- $TRINITY_HOME/Trinity.pl --seqType fq --JM 10G --left reads.ALL.left.fq --right reads.ALL.right.fq --SS_lib_type RF --CPU 6 --seqType fq —-output ./trinity_out_dir
输出文件:Trinity.fasta
4:拼接统计
- $TRINITY_HOME/util/TrinityStats.pl trinity_out_dir/Trinity.fasta>./assembly_report.txt
输出文件:assembly_report.txt
5:比对reads评估表达量(每个样本都需要单独比对)
- $TRINITY_HOME/util/align_and_estimate_abundance.pl --transcripts Trinity.fasta --seqType fq --left reads_1.fq --right reads_2.fq --est_method RSEM --aln_method bowtie --trinity_mode —prep_reference
比对输出:bowtie.csorted.bam
RSEM输出:
RSEM.isoforms.results : EM read counts per Trinity transcript
RSEM.genes.results : EM read counts on a per-Trinity-component (aka... gene) basis, gene used loosely here.
过滤比对:
- $TRINITY_HOME/util/filter_fasta_by_rsem_values.pl --rsem_output=/path/to/RSEM.isoforms.results[,...] --fasta=/path/to/Trinity.fasta --output=/path/to/output.fasta --fpkm_cutoff=1200
过滤值需要根据需求自己设定。
6:差异表达分析(edgeR)
假定有四个样本,转录本定量输出为:
LOG.isoforms.results
DS.isoforms.results
HS.isoforms.results
PLAT.isoforms.results
注意:--samples_file为样本分组信息文件 group.txt ,例如:
Throat sample2.sam
Saliva sample3.sam
Throat sample4.sam
Vaginal sample5.sam
--contrasts 为样本不同条件下比较compare.txt:
Throat Saliva
Vaginal Saliva
Throat Vaginal
7:提取最好的OFR
- $TRINITY_HOME/trinity-plugins/transdecoder/TransDecoder -t transcripts.fasta -m 100 —search_pfam /path/to/pfam_db.hmm to search —CPU 6
输出文件:
- Trinity.fasta.transdecoder.pep
- Trinity.fasta.transdecoder.cds
- Trinity.fasta.transdecoder.bed
- Trinity.fasta.transdecoder.gff3
8:功能注释
下载的软件:Trinotate、Trinity、sqlite、NCBI Blast、HMMER、signalP v4、tmhmm v2、RNAMMER
比对数据库:SwissProt、Uniref90、Pfam domains
标准化数据:
- makeblastdb -in uniprot_sprot.fasta -dbtype prot
- makeblastdb -in uniref90.fasta -dbtype prot
- hmmpress Pfam-A.hmm
blast比对(比对的数据库可以换成nr/Uniref90)
# search Trinity transcripts
- blastx -query Trinity.fasta -db uniprot_sprot.fasta -num_threads 8 -max_target_seqs 1 -outfmt 6 -evalue 1e-5 > blastx.outfmt6
# search Transdecoder-predicted proteins
- blastp -query transdecoder.pep -db uniprot_sprot.fasta -num_threads 8 -max_target_seqs 1 -outfmt 6 -evalue 1e-5 > blastp.outfmt6
功能域
- hmmscan --cpu 8 --domtblout TrinotatePFAM.out Pfam-A.hmm transdecoder.pep > pfam.log
信号肽
- signalp -f short -n signalp.out transdecoder.pep
跨膜结构
- tmhmm --short < transdecoder.pep > tmhmm.out
识别rRNA
- $TRINOTATE_HOME/util/rnammer_support/RnammerTranscriptome.pl --transcriptome Trinity.fasta --path_to_rnammer /usr/bin/software/rnammer_v1.2/rnammer
输出:Trinity.fasta.rnammer.gff
9:Load transcripts and coding regions
- $TRINITY_HOME/util/support_scripts/get_Trinity_gene_to_trans_map.pl Trinity.fasta >Trinity.fasta.gene_trans_map
- Trinotate Trinotate.sqlite init --gene_trans_map Trinity.fasta.gene_trans_map --transcript_fasta Trinity.fasta --transdecoder_pep transdecoder.pep
10:Output an Annotation Report
- Trinotate Trinotate.sqlite LOAD_swissprot_blastp blastp.outfmt6
- Trinotate Trinotate.sqlite LOAD_swissprot_blastx blastx.outfmt6
- Trinotate Trinotate.sqlite LOAD_pfam TrinotatePFAM.out
- Trinotate Trinotate.sqlite LOAD_tmhmm tmhmm.out
- Trinotate Trinotate.sqlite LOAD_signalp signalp.out
- Trinotate Trinotate.sqlite report >trinotate_annotation_report.xls
输出文件:trinotate_annotation_report.xls
原文来自:http://blog.sina.com.cn/s/blog_83f77c940102v7xu.html