

1. GCE 简介
GCE(Genome Characteristics Estimation) 是华大基因用于基因组评估的软件,其参考文献为:Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects。下载地址:ftp://ftp.genomics.org.cn/pub/gce。
GCE 软件包中主要包含 kmer_freq_hash 和 gce 两支程序。前者用于进行 kmer 的频数统计,后者在前者的结果上进行基因组大小的准确估算。
2. GCE 下载和安装
- $ wget ftp://ftp.genomics.org.cn/pub/gce/gce-1.0.0.tar.gz
- $ tar zxf gce-1.0.0.tar.gz -C /opt/biosoft
3. kmer_freq_hash 的使用
kmer_freq_hash 的常用例子:
- $ /opt/biosoft/gce-1.0.0/kmerfreq/kmer_freq_hash/kmer_freq_hash \
- -k 21 -l reads.list -a 10 -d 10 -t 24 -i 50000000 -o 0 -p species &> kmer_freq.log
kmer_freq_hash 的常用参数:
- -k
- 设置 kmer 的大小。该值为 9~27,默认值为 17 。
- -l string
- list文本文件,其中每行为一个fastq文件的路径。
- -t int
- 使用的线程数,默认为 1 。
- -i int
- 初始的 hash 表大小,默认为 1048576。该值最好设置为 (kmer 的种类数 / 0.75)/ 线程数。如果基因组大小为 100M,测序了 40M 个 reads,reads 的长度为 100bp,测序错误率为 1%,kmer的大小为 21,则kmer的种类数为100M 40M*100*1%*21=940M,若使用24线程,则该参数设置为 i=940M/0.75/24=52222222。
- -p string
- 设置输出文件的前缀。
- -o int
- 是否输出 k-mer 序列。1: yes, 0: no,默认为 1 。推荐选 0 以节约运行时间。
- -q int
- 设置fastq文件的phred格式,默认为 64。该值可以为 33 或 63。
- -c double
- 设置k-mer最小的精度,该值位于 0~0.99,或为 -1。 -1 表示不对 kmer进行过滤。设置较高的精度,可以用于过滤低质量 kmer。精度是由 phred 格式的碱基质量计算得来的。
- -r int
- 设置获取 k-mer 使用到的 reads 长度。默认使用 reads 的全长。
- -a int
- 忽略read前面该长度的碱基。
- -d int
- 忽略read后面该长度的碱基。
- -g int
- 设置使用该数目的碱基来获取 k-mers,默认是使用所有的碱基来获取 k-mer。
kmer_freq_hash 的主要结果文件为 species.freq.stat。该文件有 2 列:第1列是kmer重复的次数,第二列是kmer的种类数。该文件有255行,第225行表示kmer重复次数>=255的kmer的总的种类数。该文件作为 gce 的输入文件。
kmer_freq_hash 的输出到屏幕上的信息结果保存到文件 kmer_freq.log 文件中。该文件中有粗略估计基因组的大小。其中的 Kmer_individual_num 数据作为 gce 的输入参数。
4. gce 的使用
gce 的常用例子:
- $ /opt/biosoft/gce-1.0.0/gce \
- -f species.freq.stat -c 85 -g 4112118028 -m 1 -D 8 -b 1 > species.table 2> species.log
gce 的常用参数:
- -f string
- kmer depth frequency file
- -c int
- kmer depth frequency 的主峰对应的 depth。gce 会在该值附近找主峰。
- -g int
- 总共的 kmer 数。一定要设定该值,否则 gce 会直接使用 -f 指定的文件计算 kmer 的总数。由于默认下该文件中最大的 depth 为 255,因此,软件自己计算的值比真实的值偏小。同时注意该值包含低覆盖度的 kmer。
- -M int
- 支持最大的 depth 值,默认为 256 。
- -m int
- 估算模型的选择,离散型(0),连续型(1)。默认为 0,对真实数据推荐选择 1 。
- -D int
- precision of expect value,默认为 1。如果选择了 -m 1,推荐设置该值为 8。
- -H int
- 使用杂合模式(1),不使用杂合模式(0)。默认值为 0。只有明显存在杂合峰的时候,才选择该值为 1 。
- -b int
- 数据是(1)否(0)有 bias。当 K > 19时,需要设置 -b 1 。
gce 的结果文件为 species.table 和 species.log 。species.log 文件中的主要内容:
- raw_peak now_node low_kmer now_kmer cvg genome_size a[1] b[1]
- 84 35834245 22073804 4044916750 84.6637 4.83093e 07 0.928318 0.637648
- raw_peak: 覆盖度为 84 的 kmer 的种类数最多,为主峰。
- now_node: kmer的种类数。
- low_kmer: 低覆盖度的 kmer 数。
- now_kmer: 去除低覆盖度的 kmer 数,此值 = (-g 参数指定的总 kmer 数) - low_kmer 。
- cvg:估算出的平均覆盖度
- genome_size:基因组大小,该值 = now_kmer / cvg 。
- a[1]: 在基因组上仅出现 1 次的 kmer 之 种类数比例。
- b[1]: 在基因组上仅出现 1 次的 kmer 之 数量比例。该值代表着基因组上拷贝数为 1 的序列比例。
如果使用 -H 1 参数,则会得额外得到如下信息:
- for hybrid: a[1/2]=0.223671 a1=0.49108
- kmer-species heterozygous ratio is about 0.125918
- 上面结果中,0.125918 是由 a[1/2] 计算出来的。 0.125918 = a[1/2] / ( 2- a[1/2] ) 。
- a[1/2]=0.223671 表示在所有的 uniqe kmer 中,有 0.223671 比例的 kmer 属于杂合 kmer 。
- 此外,有 a[1/2] 和 b[1/2] 的值在最后的统计结果中。重复序列的含量 = 1 - b[1/2] - b[1] 。
则杂合率 = 0.125918 / kmer_size 。 若计算出的杂合率低于 0.2%,个人认为测序数据应该是纯合的。这时候,应该不使用 -H 1 参数。使用 -H 1 参数会对基因组的大小和重复序列含量估算造成影响。
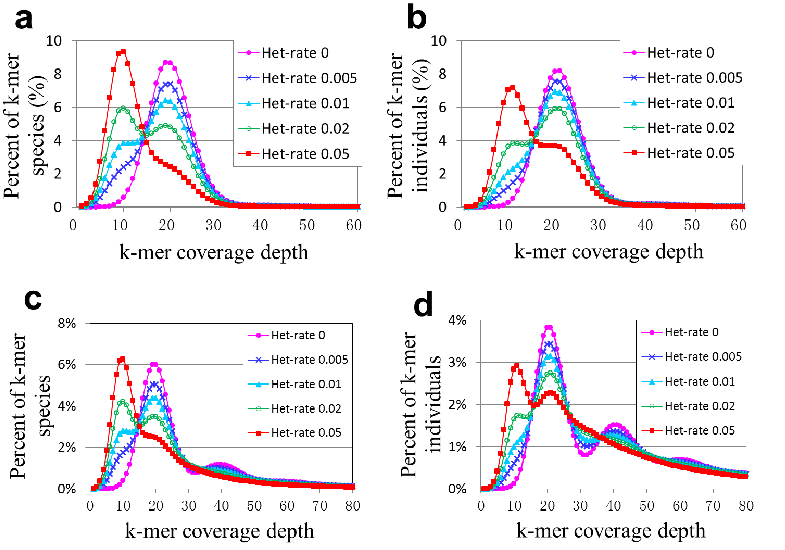
5. 不同杂合率,有无重复序列的 kmer species 和 kmer individuals 图
下图中 a 和 b 是对理想中无重复的基因组在不同杂合率下的曲线图;下图中 c 和 d 是对有重复的基因组(human)在不同杂合率下的曲线图。从下图可以参考不同杂合率下的曲线状况。
原文来自:http://www.chenlianfu.com/?p=2335
1F
请问GCE有官网和源码包吗?