

为什么要用这个软件?:因为转录组reads比对到基因组reads用bwa和bowtie的效果都不够好,所以我们选择tophat
它做了什么?:tophat把测序的转录组的原始reads比对到了参考基因组上面,并且输出了bam(二进制的sam)文件比对结果给我们。(fastq—>bam)
一:下载安装该软件
其实一般的生信服务器自然会有高手给安装好了,你只需调用即可,这里我给大家演示一下如何安装。
wget http://ccb.jhu.edu/software/tophat/downloads/tophat-2.0.13.Linux_x86_64.tar.gz
我这里简单下载二进制版本的,直接解压即可使用,里面除了tophat之外,还有好几个小工具,也是挺实用的。
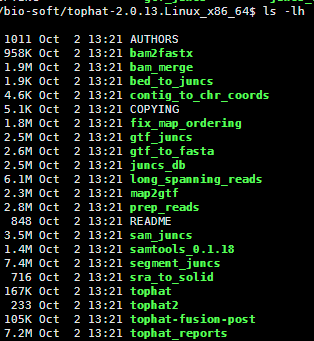
二:准备数据
数据当然是测序的转录组的原始reads啦,fastq格式的
![]()
还有人的参考基因组,这里选择人的hg19,很容易就下载了,bowtie官网里面有下载并且索引好了所有文件。如果你是自己下载的hg19,需要用bowtie先进行索引,见我的bowtie简单使用教程
三:运行命令
不需要看那么多的参数,首先要能用
/home/jmzeng/bio-soft/tophat-2.0.13.Linux_x86_64/tophat2 /home/jmzeng/ref-database/hg19 case1.fq
拆开看就是tophat2 hg19 case1.fq 即可,不需要设置任何参数,等需要优化的时候再去思考参数的意义,我这里是单端测序的命令。
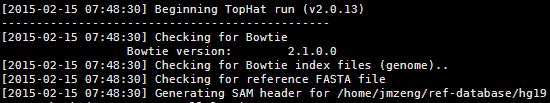
不过我的习惯是用脚本解决问题,一定要批量化运行的,我设置了30个CPU,也设置了输出目录,还加上了一个转录本
![]()
也许是要等明天才能看结果啦
好像用了30个cpu跑的很快,每个两个多小时就跑完啦

四:输出文件解读
这三个文件夹里面就输出文件,每个样本一个输出文件夹,
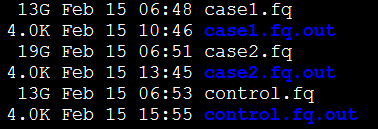
任意进入一个文件夹可以看到
通常,我们只需要那个accepted_hits.bam文件,是我们的测序reads成功比对到hg19什么的比对情况。
接下来就可以用很多软件来处理这个bam文件了
原文来自:http://www.bio-info-trainee.com/156.html