

cufflinks套装有很多,我们主要使用的只有三个
Cufflinks是用来处理tophat的输出的bam文件然后输出gtf文件
cuffmerge把多个样本的gtf文件合并的,也没啥子用,主要是测多个样本可能会需要
cuffdiff算出分组的bam文件里面的差异基因。
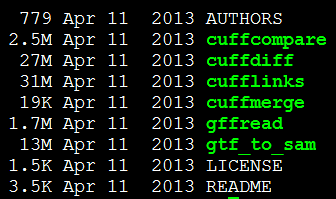
一:下载安装该软件
是二进制版本,找到网址,然后用wget下载,解压即可使用
二:准备数据
cufflinks和cuffmerge和cuffdiff需要的文件各不相同
对cufflinks来说,需要的是tophat输出的bam文件
对cuffmerge来说,需要的是多次运行cufflinks输出的多个gtf文件
对cuffdiff来说,需要的多个样本的tophat的bam文件
三:运行命令
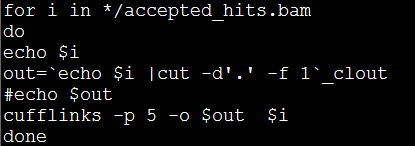
cufflinks对人类来说几乎没有用,因为对人类研究的太完全了,一大把的gtf文件,随便都可以去下载到,不需要从tophat的bam文件里面重新输出gtf文件,我简单给一个脚本
对cuffmerge来说,人类也是不需要的,主要是新物种还可以用一下,不需要脚本,就是简单的一句话而已,我都懒得讲了。
重点:对于cuffdiff来说,这个非常重要咯,我给出好几个版本的命令,希望你能总结出规律
第一个例子

有些是必须参数,跟你项目调整即可
第二个例子
cuffdiff
-o FN #这是设置结果输出目录
-b /home/immune/refer_genome/hg19/hg19 #这是参考基因组文件
-p 18 #这里cpu数量的控制
-L case,control #这是标签
-u merged_gtf/merged.gtf #这是参考gtf文件
./FN_A549_1/accepted_hits.bam,./LA_A549_2/accepted_hits.bam,./FN_A549_3/accepted_hits.bam #这是case组的bam文件地址,共三个bam文件
./FN_con_A549_1/accepted_hits.bam,./FN_con_549_2/accepted_hits.bam,./FN_con_A549_3/accepted_hits.bam #这是control组的bam文件地址,也是三个bam文件
第三个例子
![]()
对了,参考基因组还需要有个fai文件,但是cuffdiff本身也会创造,没有也无所谓的
![]()
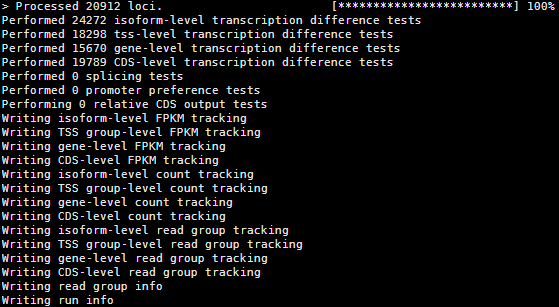
结束之后的log如下显示
4,输出数据解释,这里主要解释cuffdiff的结果文件
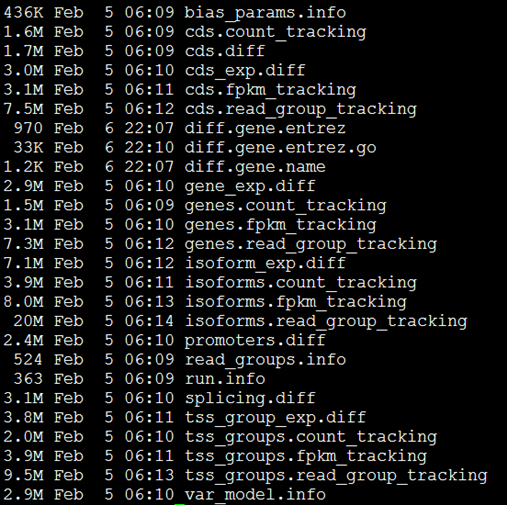
输出的文件太多了,我也没怎么具体细看。
反正那个gene_exp.diff里面可以看到差异基因的信息
主要研究了gene,isoform,cds,tss这四个方面的差异,也是我们可能会关心的四个方面的信息,同时包含着他们是如何具体算出了的tracking文件。
原文来自:http://www.bio-info-trainee.com/166.html