

edgeR是一个研究重复计数数据差异表达的Bioconductor软件包。一个过度离散的泊松模型被用于说明生物学可变性和技术可变性。经验贝叶斯方法被用于减轻跨转录本的过度离散程度,改进了推断的可靠性。该方法甚至能够用最小重复水平使用,只要至少一个表型或实验条件是重复的。该软件可能具有测序数据之外的其他应用,例如蛋白质组多肽计数数据。可用性:程序包在遵循LGPL许可证下可以从Bioconductor网站。
一:下载安装该软件
下载安装edgeR这个R包,因为这是一次讲R包的下载,我就啰嗦一点,这种生物信息学的包不同于普通的R包,是需要用biocLite来安装的,命令如下
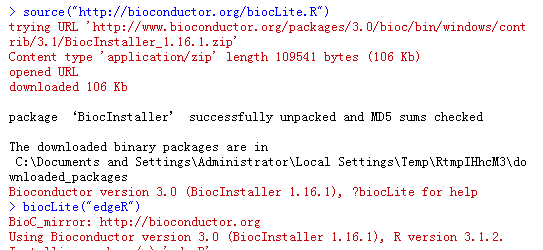
安装成功之后会有以下提示。
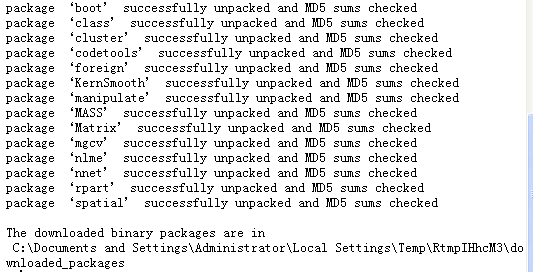
但是我加载碰到一个很幼稚的错误,因为我的电脑太差了,这是一个测试的电脑,是300块钱在二手市场里面淘的,所以内存不够。

我简单搜索了一下,才知道是虚拟内存太小了,需要调整
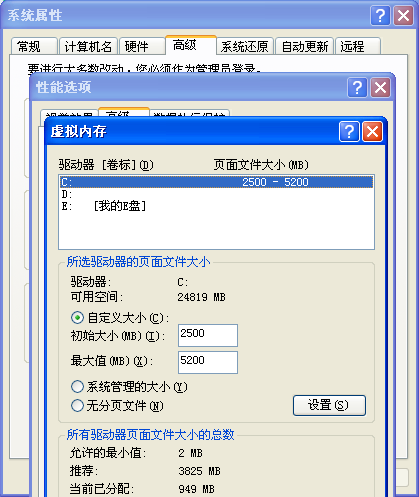
重启电脑,就成功啦
二:准备数据
就是对tophat的bam文件用HTseq计数后的count文件,见前一篇文章《转录组HTseq对基因表达量进行计数》
![]()
三:运行命令
因为主要是在R里面操作,我就只讲R里面的命令了,首先要把那些HTseq产生的文件拷贝到R的工作目录,我这里是自己设置了工作目录
setwd(“D:\\项目\\RNA-seq\\htseq”)
a=read.table(“case1.sam.count”)
b=read.table(“case2.sam.count”)
c=read.table(“control.sam.count”)
counts=data.frame(case1=a[,2],case2=b[,2],control=c[,2])
rownames(counts)=a[,1]
这样就读入了一个counts数据框
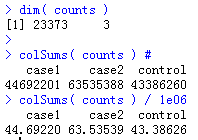
可以看到有三个样本,涉及到了23373个基因,每个样本的测序量约50M的reads
可以看到,有很多基因的计数不到30次。

我们首先对第一组来选择差异基因
case1_control=counts[,1:2];group=c(“case1″,”control”);
cds
简单看看这个构造的对象cds的具体内容

原文来自:http://www.bio-info-trainee.com/255.html