

一,所谓的网站,其实就是一个网页版的可视化软件接口而已
看看网站主页,看看它需要什么数据
http://wego.genomics.org.cn/cgi-bin/wego/index.pl
二,所需要的数据
1,human.all.go.entrez,需要自己制作,每个基因名entrez ID号,对应着一堆GO通路,人有两万多个基因,所以应该有两万多行的文件。

2,差异基因的GO通路,需要用cuffdiff得到差异基因名,然后用然后用脚本做成下面的样子。记住,上面的那个人类的背景GO文件也是一样的格式,基因名是entrez ID号,与GO通路用制表符隔开,然后每个基因所对应的GO直接用空格隔开。格式要求很准确才行。
三,上传数据,出图
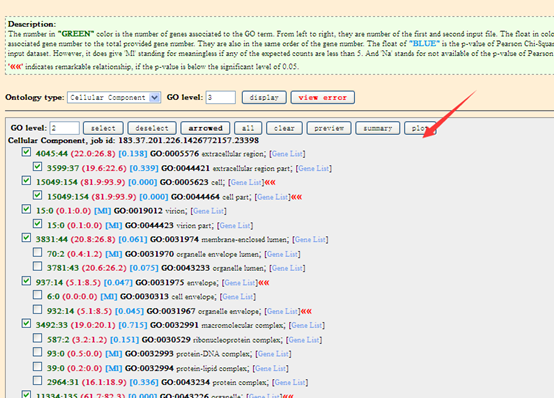
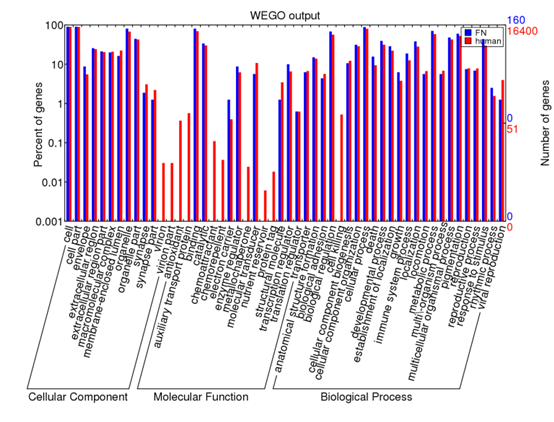
点击plot画图即可,就可以出来了一个GO通路富集图
顺便贴上wego上传数据制作的几个脚本,脚本这种东西都很难看,随便意思一下啦,用一下脚本处理就可以得到wego需要上传的数据了
1,得到差异基因名,并且转换为entrez ID号
- grep yes gene_exp.diff |cut -f 3 |sort -u >diff.gene.name
- cat diff.gene.name ../Homo_sapiens.gene_info |perl -alne ‘{$hash{$_}=1;print $F[1] if exists $hash{$F[2]}}’ |sort -u >diff.gene.entrez
2,根据找到的差异基因的entrez ID号来找到它的GO信号,输出文件给wego网站
- cat diff.gene.entrez ../gene2go |perl -alne ‘{$hash{$_}=1;print “$F[1]\t$F[2]” if exists $hash{$F[1]}}’ |perl -alne ‘{$hash{$F[0]}.=”$F[1] “}END{print “$_\t$hash{$_}” foreach keys %hash}’ >diff.gene.entrez.go
3,得到entrez ID号跟ensembl ID号的转换hash表
- perl -alne ‘{if (/Ensembl:(ENSG\d )/) {print “$1=>$F[1]”} }’ Homo_sapiens.gene_info >entrez.ensembl
4,得到人类entrez ID的go背景
- grep ‘^9606′ gene2go |perl -alne ‘{$hash{$F[1]}.=”$F[2] “}END{print “$_\t$hash{$_}” foreach sort keys %hash}’ >human.all.go.entrez
5,把人类entrez ID的go背景转换成ensembl的go背景
- cat entrez.ensembl human.all.go.entrez |perl -F”=>” -alne ‘{$hash{$F[1]}=$F[0];print “$hash{$F[0]}\t$F[1]” if exists $hash{$F[0]}}’ >human.all.go.ensembl
原文来自:http://www.bio-info-trainee.com/359.html