

以前是没有想过用这个软件的,直到有一个我的htseq无法对比对的bam文件进行基因计数(后来我才发现htseq无法计数的原因是gtf版本不同导致坐标不同,而且gtf对染色体编号没有加上chr),我简单搜索了一下,发现bedtools multicov也有类似的功能,所以我选择它来试试看!
首先注意它需要sort的bam文件及bam的index
bedtools multicov depends upon index BAM files in order to count the number of overlaps in each BAM file. As such, each BAM file should be position sorted (samtool sort aln.bam aln.sort) and indexed (samtools index aln.sort.bam) with either samtools or bamtools.
首先安装它:
wget https://github.com/arq5x/bedtools2/releases/download/v2.23.0/bedtools-2.23.0.tar.gz
解压开后
Make clean
Make all
然后就可以看到它的bin目录下全部是程序啦
命令很简单的
bedtools multicov [OPTIONS] -bams BAM1 BAM2 BAM3 … BAMn -bed <BED/GFF/VCF>
By default, multicov will report the count of alignments in each input BAM file that overlap.
例子:
bedtools multicov -bams aln1.bam aln2.bam aln3.bam -bed ivls-of-interest.bed
ivls-of-interest.bed这个文件是必须的,可能需要自己制作,其实用gtf文件也可以的
chr1 0 10000 ivl1
chr1 10000 20000 ivl2
chr1 20000 30000 ivl3
chr1 30000 40000 ivl4
输出结果前三列是坐标,第四列是基因名,跟我们的bed文件一样,只是最后三列是三个样本的计数,是添加上来的!
chr1 0 10000 ivl1 100 2234 0
chr1 10000 20000 ivl2 123 3245 1000
chr1 20000 30000 ivl3 213 2332 2034
chr1 30000 40000 ivl4 335 7654 0
同样是对gene的reads计数,bedtools的multicov工具与htseq-count的区别是
i’d guess it’s due to the fact that htseq-count only reports one hit per aligned read assuming that read is aligned uniquely and does not overlap multiple features listed in your GTF. if an aligned read hits more than one feature in your GTF then it doesn’t report that hit. bedtools gives you raw hits which includes every 1 hit for every intersection of every alignment with any features in the GTF no matter how many times it aligned or how many features it hit. you might think, “wow, htseq-count is dropping a lot of information”. yes, it is! i’ve moved to using other tools to count hits to genes (RSEM/eXpress) since they disambiguate those ambiguous alignments and as a result you get counts for all of your aligned reads. in a genome with alternative splicing you lose too much data using htseq-count, in my opinion.
而且专门有个文献在讨论这个问题
http://barcwiki.wi.mit.edu/wiki/SOPs/rna-seq-diff-expressions
http://www.nature.com/nbt/journal/v31/n1/abs/nbt.2450.html
Differential analysis of gene regulation at transcript resolution with RNA-seq
下面我讲一个实际的例子
我的bam文件如下
bedtools multicov -bams 740WT1.bam 741WT2.bam 742KO1.bam 743KO2.bam -bed mm9.bed
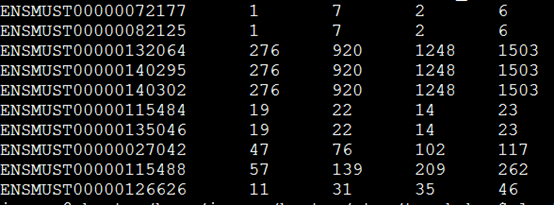
得到的这个矩阵就可以去用DESeq包来进行差异分析啦!
原文来自:http://www.bio-info-trainee.com/745.html