

一:下载安装该软件
去官网下载trinity并解压安装 http://trinityrnaseq.github.io/
安装非常简单,一个make即可
这个软件比较大,约150M。所以安装需要一会时间,以下是安装进程日志,可以看出trinity这个软件安装的同时还附带着好几个测序一起安装进来了。

安装之后即可使用啦,就在安装目录下那个绿色的就是可执行文件,可以添加到环境变量直接调用,也可以用全路径来调用。
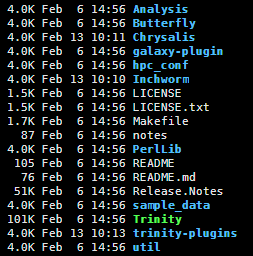
二:准备数据
它的功能就是来组装转录组的,所以需要的就是转录组的测序reads序列fastq格式即可
三:运行命令
查看help
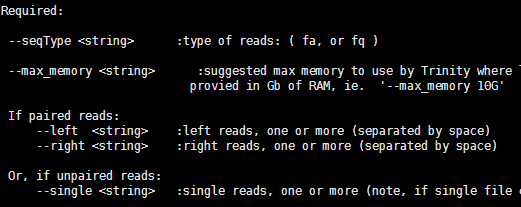
可以看到其实简单的使用trinity是非常简单的,只需要三个参数即可
- /home/jmzeng/bio-soft/trnity/Trinity –seqType fq –single SRR1793917.fastq –CPU 16 –max_memory 50G
然后就可以精心等待结果啦
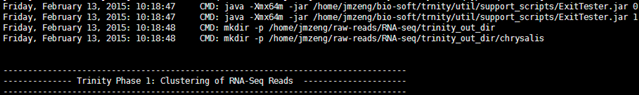
等了一晚上没看到结束,我top了一下,看到了一大堆的进程

包括了trinity的第三方插件,它的perl程序包,它的另外两个套件butterfly和chrysalis
还调用了工具包里面的一下java程序
我进去后台看了程序,第一个阶段已经完成啦,
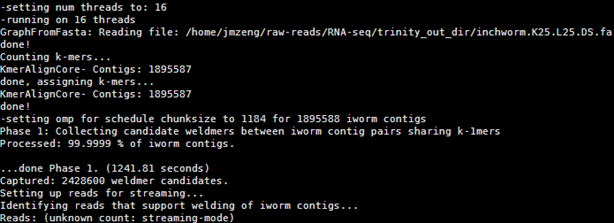
然后是第二个阶段的工作,正在进行中,第二个工作是由parafly来完成的
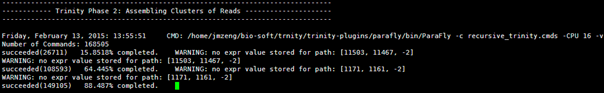
又等了一个白天,终于跑完了
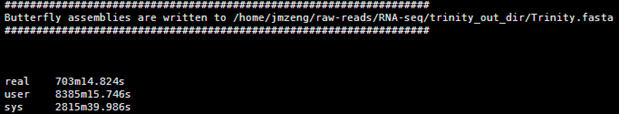
耗时约25个小时,但是我看不懂总共时间显示。
四:输出文件解读
总共产出的文件特别多
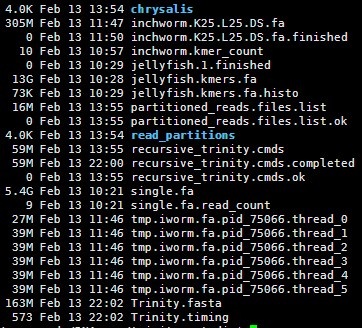
其中Trinity.fasta是最重要的, 是整个软件组装好的RNA数据
共178100条转录本
原文来自:http://www.bio-info-trainee.com/125.html