

一,下载该软件
wget http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-ubuntu64.tar.gz
tar xzf sratoolkit.current-centos_linux64.tar.gz
解压直接使用即可,里面有一大堆的软件,针对不同的测序仪,不同的数据
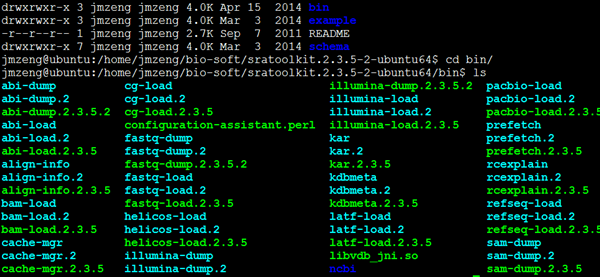
我一般只用/home/jmzeng/down_software/sratoolkit.2.3.5-2-ubuntu64/bin/fastq-dump
/home/jmzeng/down_software/sratoolkit.2.3.5-2-ubuntu64/bin/fastq-dump –split-3 SRR1793917.sra
二:下载数据
首先去NCBI里面搜索并找到你想要的数据的SRA地址,然后写脚本批量下载。
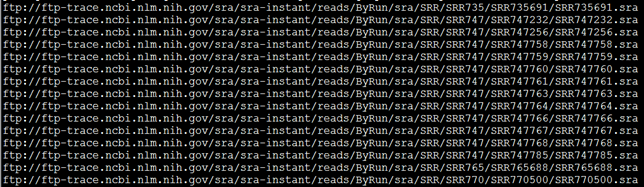
如果文献里面的SRA号,那么可以直接打开NCBI里面的搜索界面下载
如果文献里面是SRP号,那么该SRP会涉及到好几个SRA数据,得一个个开网站下载
三:用命令解压数据
下载之后的数据是

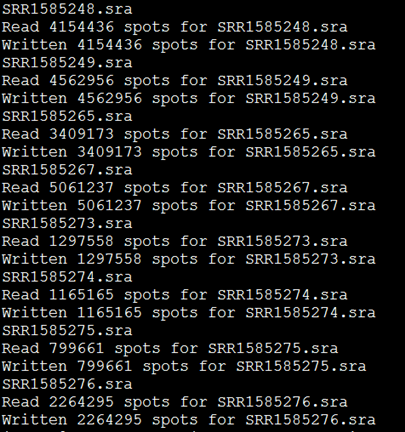
非常简单的命令,就可以把当前文件夹下的所有sra都解压开来!
- for i in *sra
- do
- echo $i
- /home/jmzeng/bio-soft/sratoolkit.2.3.5-2-ubuntu64/bin/fastq-dump --split-3 $i
- done
解压的同时它也会显示每个SRA文件的数据量
四:结果文件解读
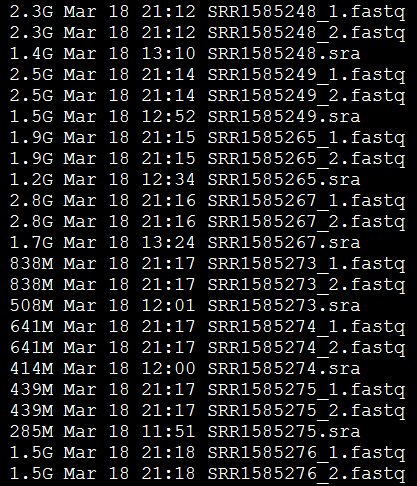
可以看到,每个SRA文件都产生了两个reads,分别是左右两端测序,说明这个SRA文件是双端测序策略。
随便打开一个fastq文件可以看到,它的读长是300bp

原文来自:http://www.bio-info-trainee.com/338.html