

一.下载该软件
http://solexaqa.sourceforge.net/index.htm
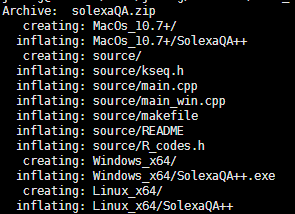
下载解压开
现在已经把它的三个功能整合到一起啦
之前是分开的程序,我主要用它的两个perl 程序,我比较喜欢之前的版本,所以下面的讲解也是基于这两个perl程序。
这两个主要是对reads进行最大子串的截取
二.准备数据。
就是我们测序得到的原始数据。
第一个就是质量控制,一般是以20为标准,当然你也可以自己设定,该软件质控的原理如下:
使用默认的参数值(defaults to P = 0.05, or equivalently, Q = 13)

基本上就是取符合阈值的最大子串。
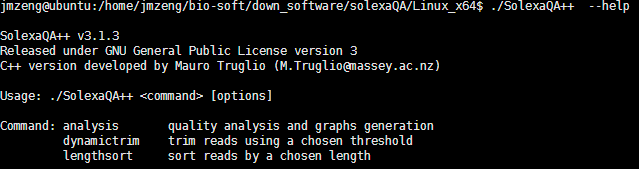
二:命令使用很简单一般使用DynamicTrim与LengthSort.pl就可以了
- for id in *fastq
- do
- echo $id
- perl DynamicTrim.pl -454 $id
- done
- for id in *trimmed
- do
- echo $id
- perl LengthSort.pl $id
- done
首先使用DynamicTrim.pl程序,非常耗时间
几个小时完毕之后
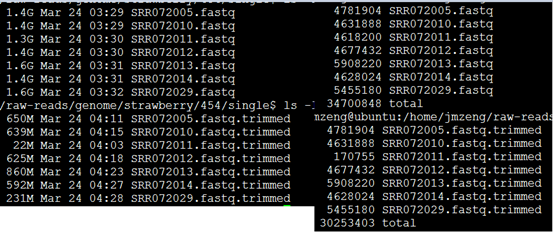
查看,产出文件如下
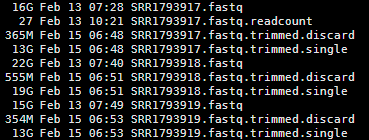
可以看到丢弃的不多,也就三五百M的
简单查看丢弃的,都是短的。
perl -lne ‘{print length if $.%4==2}’ SRR1793918.fastq.trimmed.discard |head
用这个脚本查看,可知好像都是短于25个碱基的被舍弃掉了,这个参数可以调整的。
接下来就可以用这些数据进行数据分析了
原文来自:http://www.bio-info-trainee.com/454.html