

差异分析的套路都是差不多的,大部分设计思想都是继承limma这个包,DESeq2也不例外。
DESeq2是DESeq包的更新版本,看样子应该不会有DESeq3了,哈哈,它的设计思想就是针对count类型的数据。
可以是任意features的count数据,比如对各个基因的count,或者外显子,或者CHIP-seq的一些feature,都可以用来做差异分析。
使用这个包也是需要三个数据:
- 表达矩阵
- 分组矩阵
- 差异比较矩阵
总结起来三个步骤,我下面会一一讲解
- 重点就是构造一个dds的对象,
- 然后直接用DESeq函数进行normalization处理即可,
- 处理之后用results函数来提取差异比较结果。
读取自己的数据
一般我们会自己读取表达矩阵和分组信息,下面是一个例子:
options(warn=-1) suppressMessages(library(DESeq2)) suppressMessages(library(limma)) suppressMessages(library(pasilla)) data(pasillaGenes) exprSet=counts(pasillaGenes) head(exprSet) ##表达矩阵如下 ## treated1fb treated2fb treated3fb untreated1fb untreated2fb ## FBgn0000003 0 0 1 0 0 ## FBgn0000008 78 46 43 47 89 ## FBgn0000014 2 0 0 0 0 ## FBgn0000015 1 0 1 0 1 ## FBgn0000017 3187 1672 1859 2445 4615 ## FBgn0000018 369 150 176 288 383 ## untreated3fb untreated4fb ## FBgn0000003 0 0 ## FBgn0000008 53 27 ## FBgn0000014 1 0 ## FBgn0000015 1 2 ## FBgn0000017 2063 1711 ## FBgn0000018 135 174 (group_list=pasillaGenes$condition) ## [1] treated treated treated untreated untreated untreated untreated ## Levels: treated untreated ##这是分组信息,7个样本,3个处理的,4个未处理的对照!
第一步:构建dds对象
那么根据这两个数据就可以构造dds的对象了
colData <- data.frame(row.names=colnames(exprSet),
group_list=group_list
)
## 这是一个复杂的方法构造这个对象!
dds <- DESeqDataSetFromMatrix(countData = exprSet,
colData = colData,
design = ~ group_list)
## design 其实也是一个对象,还可以通过design(dds)来继续修改分组信息,但是一般没有必要。
dds
## class: DESeqDataSet
## dim: 14470 7
## exptData(0):
## assays(1): counts
## rownames(14470): FBgn0000003 FBgn0000008 ... FBgn0261574
## FBgn0261575
## rowData metadata column names(0):
## colnames(7): treated1fb treated2fb ... untreated3fb untreated4fb
## colData names(1): group_list可以看到我们构造的dds对象有7个样本,共14470features
从基因名可以看出,是果蝇的测序数据
我们也可以直接从expressionSet这个对象构建dds对象!
library(airway) data(airway) suppressMessages(library(DESeq2)) dds <- DESeqDataSet(airway, design = ~ cell+ dex) design(dds) <- ~ dex ## 构造好的对象还可以更改分组信息
第二步:做normalization
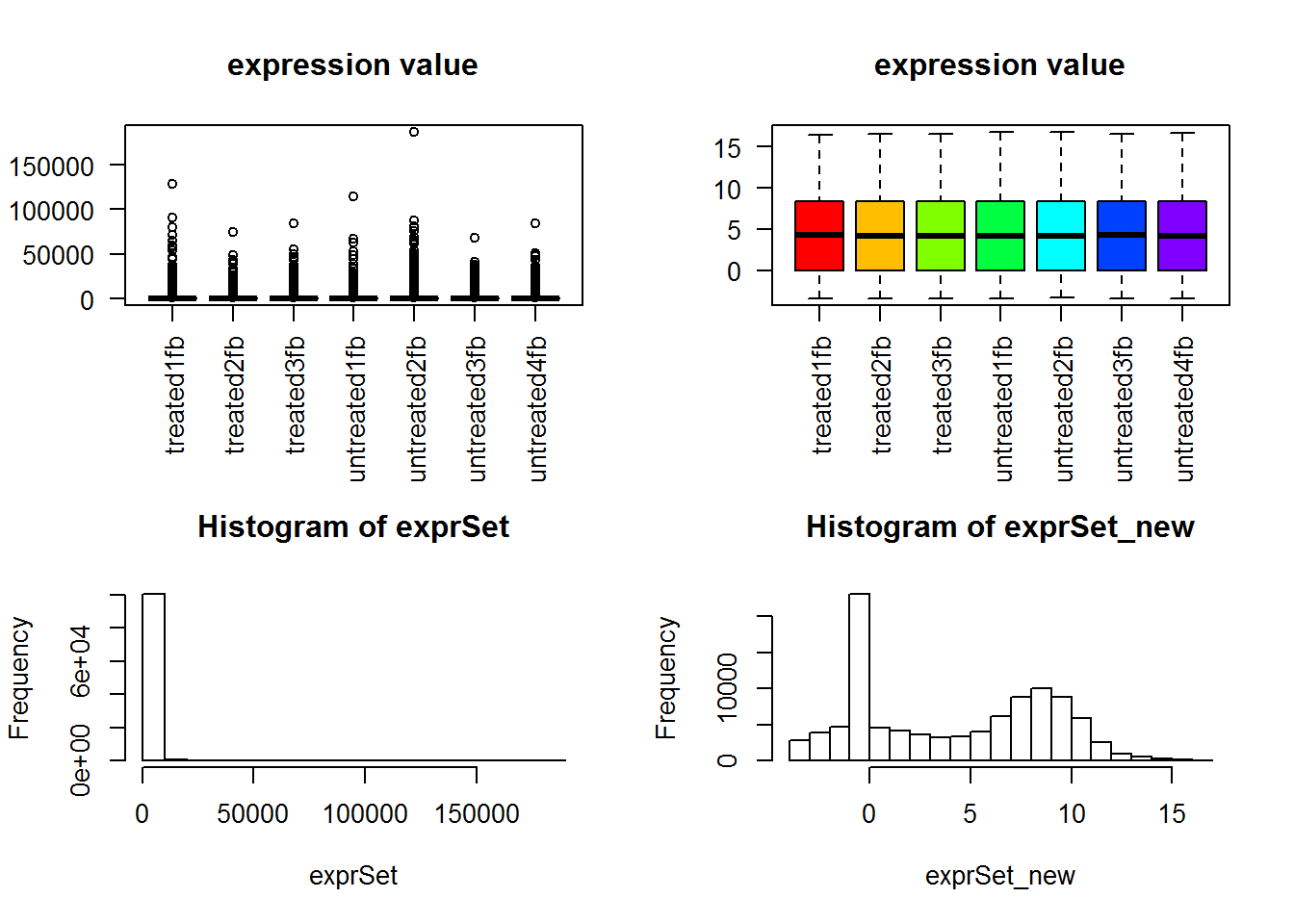
suppressMessages(dds2 <- DESeq(dds)) ##直接用DESeq函数即可 ## 下面的代码如果你不感兴趣不需要运行,免得误导你 ## 就是看看normalization前面的数据分布差异 rld <- rlogTransformation(dds2) ## 得到经过DESeq2软件normlization的表达矩阵! exprSet_new=assay(rld) par(cex = 0.7) n.sample=ncol(exprSet) if(n.sample>40) par(cex = 0.5) cols <- rainbow(n.sample*1.2) par(mfrow=c(2,2)) boxplot(exprSet, col = cols,main="expression value",las=2) boxplot(exprSet_new, col = cols,main="expression value",las=2) hist(exprSet) hist(exprSet_new)
第三步:提取差异分析结果
resultsNames(dds2)
## [1] "Intercept" "group_listtreated" "group_listuntreated"
## 其实如果只分成了两组,并没有必要指定这个比较矩阵!
res <- results(dds2, contrast=c("group_list","treated","untreated"))
## 提取你想要的差异分析结果,我们这里是trt组对untrt组进行比较
resOrdered <- res[order(res$padj),]
resOrdered=as.data.frame(resOrdered)
head(resOrdered)
## baseMean log2FoldChange lfcSE stat pvalue
## FBgn0039155 453.2753 -4.281830 0.1919977 -22.30146 3.576174e-110
## FBgn0029167 2165.0445 -2.182745 0.1080670 -20.19807 1.017931e-90
## FBgn0035085 366.8279 -2.436860 0.1505280 -16.18875 6.054219e-59
## FBgn0029896 257.9027 -2.511257 0.1823764 -13.76964 3.881667e-43
## FBgn0034736 118.4074 -3.166392 0.2375506 -13.32933 1.562878e-40
## FBgn0040091 610.6035 -1.526400 0.1278555 -11.93848 7.457520e-33
## padj
## FBgn0039155 2.764025e-106
## FBgn0029167 3.933795e-87
## FBgn0035085 1.559769e-55
## FBgn0029896 7.500351e-40
## FBgn0034736 2.415897e-37
## FBgn0040091 9.606528e-30差异分析结果很容易看懂啦!
原文来自:www.bio-info-trainee.com
1F
请问分组矩阵是一行7列吗?还是一行8列?就根据您给出的例子来说。
B1
@ Pan 我觉得你问的矩阵是一个两列的表。 第一列放样品名,另一列放入对应的分组名。
2F
请问大神,筛选差异基因的标准是什么? log2FC>=1,而且padj应该小于多少? 通常有0.05、 0.01. 我们怎么把握这个阈值的标准呢?